[Previous]
La fragilité héréditaire des nerfs périphériques à la compression : Une étude électrophysiologique et génétique
La fragilité héréditaire des nerfs périphériques à la compression, ou hereditary neuropathy with liability to pressure palsies (HNPP), a été décrite pour la première fois par De Jong en 1947 et dans la littérature anglo-saxone par Davies en 1954. C'est une neuropathie héréditaire chronique caractérisée par des épisodes récidivants de paralysie avec paresthésies. La prévalence des neuropathies héréditaires motrices et sensorielles (hereditary motor and sensory neuropathy HMNS) est estimée à 1 pour 10000 (Nelis et al., 1996).
L'HNPP se manifeste à tous les âges, le début des symptômes étant le plus souvent reconnu durant la deuxième et troisième décennie. Bien que les deux sexes puissent être concernés, on note une légère prédominance masculine, probablement liée à une exposition accrue aux risques professionnels (Meier et Moll, 1982).
La clinique est variable, les patients pouvant se présenter avec une attaque de paralysie franche ou alternativement de manière plus insidieuse. L'éventail des symptômes comprend des paresthésies et engourdissements de courte durée jusqu'à des paralysies chroniques. L'examen clinique peut révéler des réflexes tendineux diminués ou absents dans les régions concernées (Meier et Moll, 1982). L'évolution de l'affection est le plus souvent favorable bien qu'une progression plus sévère soit également décrite. Il est important que les sujets atteints comprennent leur neuropathie et puissent ainsi prendre des mesures préventives afin d'éviter les facteurs déclenchants. La décompression chirurgicale peut parfois améliorer sinon éliminer la symptomatologie (Roth et Magistris, 1984).
L'évènement qui déclenche les attaques est en général un traumatisme, une traction ou une compression d'un nerf périphérique. Certaines positions prolongées suffisent parfois à déclencher les symptômes. Parfois aucun processus initial ne peut être trouvé lors de l'anamnèse.
Les caractéristiques électrophysiologiques de l'HNPP ont été établies en grande partie par Earl et al. en 1964. On note un ralentissement des vitesses de conduction nerveuses et une réduction d'amplitude des potentiels d'actions globaux moteurs et sensitifs des nerfs atteints cliniquement. Les anomalies électrophysiologiques peuvent concerner des nerfs non atteints cliniquement ainsi que des membres asymptomatiques de la famille. Le ralentissement des vitesses de conduction est plus prononcé aux sites d'enclavements ainsi que dans les segments distaux des nerfs (Behse et al, 1972). Des signes de régénération axonale avec des réflexes d'axones moteurs sont également observés (Roth 1978, Magistris et Roth, 1985). Des blocs de conduction expliquent les épisodes de paralysie; ils sont parfois persistants (Magistris et Roth, 1985).
Histologiquement l'HNPP présente des épaississements localisés de la myéline. Décrits pour la première fois par Behse en 1972, ces épaississements ont été appelés "tomacula" par Madrid et Bradley en 1975. Bien que les épaississements de la myeline ne soient pas spécifiques de l'HNPP, leur présence en grand nombre l'est. L'amyotrophie est rare ou tardive dans l'HNPP.
Le diagnostic différentiel se fait principalement parmi les neuropathies héréditaires, dont l'amyotrophie hérédo-familiale du plexus brachial et le syndrome de Charcot-Marie-Tooth (CMT1A) sont des exemples.
L'HNPP est transmise sur un mode autosomique dominant. La recherche génétique a depuis quelques années apporté de nombreux développements dans la compréhension de cette neuropathie. L'anomalie génétique la plus souvent mise en cause est une délétion de 1.5 mégabases sur le bras court du chromosome 17. C'est une région du chromosome qui contient le gène PMP-22 (Peripheral Myelin Protein 22) (Chance et al, 1993; Mariman et al, 1993). Le locus de ce gène a été impliqué tout d'abord en utilisant des techniques de "linkage analysis" mais des mutations ponctuelles de quelques paires de bases dans l'exon 1 de ce gène ont par la suite confirmé son importance dans la pathogénie de la maladie (Nicholson et al, 1994). Des mutations ponctuelles ont depuis été mises en évidence chez des sujets qui ne présentaient pas la délétion 17p11.2 complète. Depuis ces découvertes initiales, des études cliniques et génétiques ont confirmé que la délétion 17p11.2 est l'anomalie génétique la plus courante chez les patients présentant des anomalies de conduction nerveuse et des présentations cliniques typiques et se trouve chez 85% des patients (Gouider et al, 1995, Tyson et al, 1996). D'autres auteurs ont suggéré qu'il existe une hétérogénéité génétique de l'HNPP (Mariman et al, 1994).
Le rôle exact de la protéine PMP-22 n'est pas connu. Des études montrent que la protéine est présente dans la myéline compacte et suggèrent qu'elle n'est pas seulement une protéine structurale mais qu'elle serait également un canal ou un pore non-spécifique (Suter et al, 1993). L'expression anormale du mRNA, et donc de la protéine, a été proposée comme étant le facteur déterminant dans la pathogénie moléculaire d'une série de maladies dont la maladie de Charcot-Marie-Tooth 1A, celle de Déjerine-Sottas et l'HNPP (Yoshikawa et al, 1994). En tout état de cause il est démontré que l'expression correcte de la protéine PMP-22 est essentielle à la myélinisation normale (Trapp et al, 1996; Murakami et al, 1996).
La région 17p11.2 de 1.5 Mb de l'HNPP est précisément celle qui présente une duplication dans la maladie de Charcot-Marie-Tooth Type 1A (Valentijn et al, 1992). Ceci constitue le premier exemple humain d'un syndrome résultant de produits réciproques d'échanges inégaux de segments intra-chromosomiques, et en particulier de crossover inégaux survenant durant la méiose (Chance et al, 1994). D'autres mécanismes qui pourraient produire la délétion ont été proposés. Le Guern et al, 1996, ont suggéré que la délétion pouvait être le résultat de réarrangements intra-chromosomiques dans des cas de novo d'origine maternelle. Il est remarquable que le résultat de la duplication soit similaire à celui de la délétion puisque la CMT fait partie du diagnostic différentiel de l'HNPP (Roth et Magistris, 1984).
Au vu de l'hétérogénéité, cette recherche vise à vérifier si les patients suivis dans l'unité d'électroneuromyographie et des affections neuromusculaires de la clinique de Neurologie de l'Hôpital Cantonal, pour une HNPP basé sur des observations cliniques et électrophysiologiques, sont porteur de la délétion 17p11.2. Vingt quatre individus appartenant à 23 familles on été étudiés. Quatres domaines cliniques, soit l'anamnèse personnelle et familiale, les observations cliniques et l'étude électrophysiologique, ont été utilisés pour déterminer une probabilité d'HNPP, et chaque individu inclu dans l'étude a été classé dans un groupe de faible, moyenne, ou forte probabilité. Ceci a été comparé au résultat génétique. La technique du Fluorescent in situ hybridisation (FISH) et le Southern blotting quantitatif sont celles qui ont été utilisées pour l'analyse génétique. La délétion 17p11.p a été trouvée chez 7 individus appartenant à 6 familles. Parmi ces individus, 4 étaient fortement suspects de présenter une HNPP. Dans une famille, 2 des 3 membres étudiés présentaient la délétion. Pris séparément, les différents composants du score proposé sont mal corrélés avec les résultats génétiques. En score composé par contre, la corrélation est bonne, la délétion ayant été retrouvée chez tous les sujets présentant une forte probabilité.
Un individu présentant a) une neuropathie transmise sur un mode autosomale dominante, b) avec des plaintes de parésie et paresthésies chroniques et récurrentes à mettre en relation avec des traumatismes mineurs, c) et chez qui l'étude électrophysiologique montre un ralentissement des vitesses de conduction nerveuses et des blocs de conduction, a une grande probabilité d'être atteint de l'HNPP. La délétion 17p11.2 est spécifique de l'HNPP et permet donc comme une confirmation du diagnostique.
Aujourd'hui, diverses techniques de biologie moléculaire permettent de détecter la délétion 17p11.2, qui constitue l'anomalie génétique la plus fréquente de l'HNPP. Au vu de la diversité de la génétique de l'HNPP, les techniques qui ne détectent que la délétion n'excluent pas des mutations ponctuelles dans le gène PMP-22; seules les techniques qui permettent une évaluation directe de la séquence d'ADN peuvent exclure toute anomalie.
Il n'y a pas de traitement de l'HNPP mais la confirmation du diagnostic est en soi souvent importante pour le patient et permet la mise en oeuvre de moyens de prévention simples. Nous disposons aujourd'hui d'un test moléculaire qui allie efficacité à des coûts acceptables, qui permet une confirmation diagnostique rapide. Les tests génétiques sont particulièrement utiles chez les individus ayant une présentation clinique et électrophysiologique atypique. La confirmation du diagnostic génétique permet un conseil génétique pour l'individu et les membres de sa famille. Les sujets atteints doivent éviter des mouvements, postures ou situations favorisant le traumatisme d'un nerf. Cette étude ne permet pas de conclure à une possible hétérogénéité génétique de l'HNPP mais elle établit une base pour procéder à des tests plus approfondis avec recherche de mutations du gène PMP22.
1. Summary
1.1 Abstract
Hereditary neuropathy with liability to pressure palsies (HNPP) is a chronic peripheral nerve disorder. Individuals typically present chronic and recurrent complaints of palsies with paraesthesiae related to minor trauma. Electrophysiological studies show nerve conduction slowings and conduction blocks, usually more marked at entrapment sites. Schwann cell swellings ('tomacula') are morphological changes of peripheral nerves characteristic of HNPP. A 1.5 Mb deletion on chromosome 17p11.2 is considered specific of HNPP and serves as definite confirmation of the diagnosis. The implicated gene is PMP22, which codes for a peripheral myelin protein. This research aims to determine whether patients currently being followed for symptoms and complaints consistent with HNPP and having electrophysiological studies suggestive of HNPP, actually have the implicated deletion. The individuals enrolled in this study were those being followed by the ENMG unit of the HUG Neurology Clinic. Twenty-four subjects from 22 families were studied. The individuals' clinical and electrophysiological findings were used to determine their respective clinical probability of having HNPP. Four areas of particular value were used for the proposed clinical scoring system, namely personal and family history, clinical and electrophysiological findings. The main diagnostic technique used to detect the 1.5 Mb PMP22 deletion on chromosome 17 was fluorescent in situ hybridisation (FISH). The deletion was found in 7 of the 24 individuals studied. Of these 7 patients, 4 were highly suspected of having HNPP on the basis of the clinical and electrophysiological findings. The deletion was found in 3 individuals in whom HNPP was not strongly suspected, and notably in one whose findings suggested a low probability. In one family, of the 3 members investigated, 2 had the deletion. Personal and family history, and clinical findings, taken individually, were poorly correlated with the genetic results. Taken together as composite score however, the deletion was found in all the patients with a high probability. Although there is presently no curative treatment for this neuropathy, providing a diagnosis to patients is of interest since simple preventive measures to avoid nerve trauma can greatly reduce the potential disability associated with HNPP. With the advent of modern molecular analysis, rapid and effective diagnosis is becoming a reality. Genetic testing is of particular interest in patients with an atypical clinical and electrophysiological presentation. Confirming the presence of the genetic defect has implications for the individual as well as for family members, allowing for preventive measures and genetic counselling. Our results do not provide a conclusive answer to the question of a possible genetic heterogeneity. This study lays the groundwork for further investigations to be carried out. In particular direct sequence analysis could provide interesting insights into the disease and its presentation.
1.2 Résumé
La fragilité héréditaire des nerfs périphériques à la compression (HNPP) est une neuropathie périphérique chronique caractérisée par des épisodes récidivants de paralysie avec paresthésies. L'événement qui déclenche les attaques est en général un traumatisme ou la compression d'un nerf. Les études électrophysiologiques montrent un ralentissement des vitesses de conduction nerveuses, ainsi que des blocs de conduction, prédominants aux lieux d'enclavements. A l'histologie, l'HNPP présente des épaississements localisés de la myéline appelés "tomacula". L'HNPP est transmise sur un mode autosomique dominant. L'anomalie génétique la plus souvent mise en cause est une délétion de 1.5 mégabases sur le bras court du chromosome 17. C'est une région du chromosome qui contient le gène PMP22 qui code pour une protéine de la myéline périphérique. Au vu de l'hétérogénéité de présentation, cette étude a visé à vérifier si les patients suivis par la Clinique de Neurologie de l'Hôpital Cantonal pour une HNPP suspectée sur les observations cliniques et électrophysiologiques, sont porteurs de la délétion 17p11.2. Vingt-quatre individus appartenant à 22 familles ont été étudiés. Quatre domaines, soit l'anamnèse personnelle et familiale, les trouvailles cliniques et l'étude électrophysiologique, ont été utilisés pour déterminer une probabilité d'HNPP, et chaque individu inclus dans l'étude a été classé dans un groupe de faible, moyenne ou forte probabilité. Ceci a été comparé au résultat génétique. La Fluorescent in situ hybridisation (FISH) est la technique principalement utilisée pour l'analyse génétique. La délétion 17p11. a été trouvée chez 7 individus appartenant à 6 familles. Parmi ces individus, 4 étaient fortement suspect de présenter une HNPP. La délétion a également été trouvée chez 2 individus chez qui la suspicion était respectivement moyenne et faible. Dans une famille, 2 des 3 membres étudiés présentaient la délétion. Pris séparément, les composants du score proposé sont mal corrélés avec les résultats génétiques. En score composé par contre, la corrélation est bonne, la délétion ayant été retrouvée chez tout les sujets présentant une forte probabilité. Il n'y a pas de traitement de l'HNPP mais la confirmation du diagnostic est en soi souvent importante pour le patient et permet la mise en oeuvre de moyens de prévention simples. Nous disposons aujourd'hui d'un test moléculaire qui permet une confirmation diagnostique rapide. Les tests génétiques sont particulièrement utiles chez les individus ayant une présentation clinique et électrophysiologique atypique. La confirmation du diagnostic génétique permet un conseil génétique pour l'individu et les membres de sa famille. Cette étude ne permet pas de conclure concernant une possible hétérogénéité génétique de l'HNPP mais elle établit une base pour procéder à des tests plus approfondis avec recherche de mutations du gène PMP22.
2. Introduction
2.1 Historical background, clinical presentation and evolution
Hereditary neuropathy with liability to pressure palsies (HNPP) is a chronic peripheral nerve disorder characterised by acute and often recurrent palsies with paraesthesiae. The earliest descriptions of the condition were made by De Jong in 1947, and by Davies in 1954. These authors reported several cases in families with recurrent peripheral nerve palsies. Wahle and Tönnis first used the term HNPP in 1958. Since then the disorder has been progressively better characterized. The prevalence of hereditary motor and sensory neuropathies (HMSN), of which HNPP constitutes an unclear proportion, is estimated at 1 in 10000 (Nelis et al., 1996).
The onset of HNPP can occur at any age, but the disorder often starts during the second to third decade of life. HNPP affects both males and females, but there is a slightly higher incidence in males; this may be due to their greater exposure to occupational hazards (Meier and Moll, 1982). The clinical presentation of HNPP varies; it sometimes presents in a dramatic manner with outright paralysis or alternatively appears rather insidiously. Symptoms range from short-lived paraesthesiae and numbness, to chronic paralysis. Fasciculations and myokymias in certain muscle groups have also been described as presenting symptoms (Andreadou et al., 1995). Clinical examination sometimes reveals decreased or absent deep tendon reflexes in given areas (Meier and Moll, 1982; Serratrice et al., 1987). Severe and incapacitating evolution of the disease has been described, but is rare (Barbieri et al., 1990). Pain is unusual but when present it usually involves the brachial plexus (Meier and Moll, 1982).
The triggering event to HNPP is generally trivial and seemingly minor trauma, traction or compression of a particular peripheral nerve. In some instances no initiating process can be found. Typically the prolonged exposure of some nerves to certain positions may suffice to produce symptoms (Meier and Moll, 1982). The brachial plexus, for example, can be injured whilst lying on one side, sleeping with an arm above the head, or carrying a heavy weight. Sitting with crossed legs can imply the common peroneal nerve. Nerves are especially susceptible at entrapment sites; the ulnar nerve at the elbow is a common example of this. Other sites in the upper limb include the median nerve in the carpal tunnel, the ulnar nerve in the Guyon canal, and the radial nerve when it enters the anterior region of the arm. In the lower limb the most common impairment is the common peroneal nerve at the head of the fibula. In their review, Meier and Moll analysed 290 attacks in 93 patients and showed that the peroneal and ulnar nerves were the most frequently involved, being affected in 35% and 20% of the cases respectively (Meier and Moll, 1984, Figure 1).
 Fig. 1 Analysis of 290 nerve palsies in 93 patients (taken from well documented attacks in literature and from investigated series).
Reproduced from Meier and Moll, 1982.
Fig. 1 Analysis of 290 nerve palsies in 93 patients (taken from well documented attacks in literature and from investigated series).
Reproduced from Meier and Moll, 1982.
 Fig. 2 Age distribution of index cases of 40 HNPP families (80 patients).
Reproduced from Meier and Moll, 1982.
Fig. 2 Age distribution of index cases of 40 HNPP families (80 patients).
Reproduced from Meier and Moll, 1982.
The evolution of HNPP is extremely varied. Earl et al. (1964) in their study of four families showed that most incidents involving palsies are followed by recovery, occasionally incomplete. Roth and Magistris (1984) reported cases of unremitting palsies that lasted for years. The progression of the disease is closely linked to the manner in which patients adapt to the condition. Presently the best course of action is for a patient to understand and therefore avoid movements, postures or situations that act as triggering factors. In some cases surgical decompression of nerves, or neurolysis, has proved effective in alleviating or eliminating symptoms (Roth and Magistris, 1984).
2.2 Electrophysiological aspects
The electrophysiological findings in HNPP have been comprehensively established. Earl et al. (1964) were the first to publish a systematic study of the problem. They showed that there is a general slowing of nerve conduction velocities and a reduction of the amplitude of the compound motor and sensory action potentials in clinically affected nerves. It is interesting to note that electrophysiological abnormalities are also found in clinically unaffected nerves, as well as in asymptomatic members of families with HNPP. Other particularities that have been reported involve slowing of conduction velocities, especially at entrapment sites and in distal segments of nerves, and the fact that sensory nerve fibres are more consistently affected than motor nerve fibres (Behse et al., 1972). In 1985 Magistris and Roth published a report of long lasting conduction blocks found in patients with HNPP. The multiplicity and chronicity of the blocks at entrapment sites was considered characteristic. A total of 38 patients were examined and extensive electrophysiological investigations were carried out. Roth was the first to describe conduction blocks of long duration that were directly related to this particular disease (Roth, 1978). Since then other conditions leading to persistent conduction blocks have been defined: Multifocal Sensory Motor Neuropathy (Lewis et al., 1982), Multifocal Motor Neuropathy (Roth et al., 1986), and nerve lesions that follow radiotherapy (Roth et al., 1988).
Interestingly, electrophysiological studies have shown that motor axon reflexes (MAR) are found in patients with HNPP. MARs are electrophysiological responses found when damaged peripheral axons regenerate and give birth to more than one axonal extension. Motor axon reflex potentials are electrophysiological responses that have distinctive characteristics. The observed presence of MARs at first electrophysiological examination demonstrates that the neuropathy in HNPP brings about chronic and long lasting axonal regeneration (Roth, 1978; Roth and Magistris, 1984; Magistris and Roth, 1985).
2.3 Pathology and histological aspects
Morphological changes are often found in HNPP, and nerve biopsies are particularly revealing. Behse et al. (1972) were the first to describe local thickenings of the myelin sheath around axonal fibres whilst studying the sural nerve. These Schwann cell swellings were termed "tomacula", from the latin word for sausage, by Madrid and Bradley (1975). Taking into account this histological characteristic, the term "tomaculous neuropathy" is used by certain authors to designate this condition. The focal swellings of the nerve fibres consist of an excessive number of abnormally folded myelin lamellae. Behse et al. (1972) in their initial work, showed that the swellings consisted of over 500 myelin lamellae, whereas normal nerve samples have between 90 and 200. Most authors who have found and described tomacula have used a method known as "teasing", whereby single fibres are isolated and then examined individually (Behse et al., 1972). Oda et al. (1990) showed that the tomacula tended to line up on certain axons instead of being distributed randomly. These authors suggested that this finding indicates that, in addition to the genetically determined generalized myelination disorder, some signals emanating from the axons might play a role in the formation of tomacula. Focal thickening of myelin is not specific to HNPP and has been observed in neuropathies such as Charcot-Marie-Tooth disease and DejerineSottas disease (Pouget et al., 1992). Nevertheless it is only in HNPP that tomacula are found in such abundance (Pouget et al., 1992). Behse et al. (1972), for example, counted 1 per 200-300 nerve fibres in random crosssection cuts, and showed that one fourth of the internodal segments in their study contained portions in which the diameter was increased to as much as twice that of the remaining segment. The swellings were located in both nodal and paranodal segments. Within these areas the axonal diameter is reduced and Behse et al. (1972) suggested that this change in the axonal to myelin ratio could account for some of the slowing of conduction velocities.
Several mechanisms have been proposed for the formation of tomacula (Behse et al., 1972; Meier and Moll, 1982). One theory holds that they are a result of hypermyelination with excessive numbers of myelin lamellae being deposited in a periodic fashion around the axon. However, tomacula most frequently seem to result from an abnormal folding of the myelin lamellae; the formation of redundant loops with secondary wrapping of double-folded loops around the axon creating asymmetric thickening of the myelin sheath.
Other findings such as focal demyelination and remyelination, loss of large myelinated fibres and an increase in number of small myelinated fibres have been reported (Behse et al., 1972; Meier and Moll, 1982). In a 1984 article reporting 23 patients, Roth and Magistris suggested that the conduction blocks mentioned earlier could be caused by the influence of the tomacula on the nodes of Ranvier as well as by the frequently accompanied intussusception of one myelin segment into a neighbouring segment with consecutive displacement of the node. Only a few nerve biopsies were carried out in this study because clinical and electrophysiological findings were considered to suffice for the diagnosis (Roth and Magistris, 1984; Magistris and Roth, 1985). Significantly, these authors mentioned a number of instances in which tomacula were not found, or were rare, in clinically typical cases of HNPP; other authors have also reported this (Behse et al., 1972; Windebank, 1984). It has been suggested that the absence of tomacula in some cases indicates that the focal thickening of myelin might only be present at certain stages of the pathological process (Behse et al., 1972; Windebank, 1984). That tomacula have not been found in great numbers in all individuals with HNPP diagnosed on the basis of clinical and electrophysiological criteria is suggestive of an underlying genetic heterogeneity. It will be discussed later.
Amyotrophy is unusual at the time of the first episodes of HNPP (Pouget et al., 1992). With repeated lesions and paralysis muscle atrophy eventually occurs certain muscle groups of the foot as well as those innervated by the ulnar nerve are reportedly most at risk (Pouget et al., 1992). Magistris and Roth (1985) have suggested that muscle atrophy is not consistently found because axoplasmic transport of trophic factors remains unaffected even in the presence of conduction blocks.
A

B
 Fig. 3, A and B (X160 and X400 respectively) Pictures showing tomacula in myelin sheath of individual axons (X160). A method known as "teasing" is used to obtain the single fibres.
Pictures by Dr GP Pizzolato, Unité de neuropathologie, département de pathologie clinique, Genève.
Fig. 3, A and B (X160 and X400 respectively) Pictures showing tomacula in myelin sheath of individual axons (X160). A method known as "teasing" is used to obtain the single fibres.
Pictures by Dr GP Pizzolato, Unité de neuropathologie, département de pathologie clinique, Genève.
2.4 Diagnosis of HNPP
Diagnosis of HNPP requires a multidisciplinary approach. Some of the most essential diagnostic information comes from the family history of a patient, with, whenever possible, special importance given to establishing an accurate family tree. In this way an evaluation of the type of genetic transmission can be established even if the heterogeneity of the clinical presentation may make this difficult. The age of onset and precise conditions under which the symptoms present have to be determined, and the notion of relapsing or recurrent episodes is important. Electrophysiological studies with investigation of nerve conduction velocities and the search for conduction blocks are the next step. Electrophysiological studies of family members may be used to demonstrate that the neuropathy under study is hereditary. Nerve conduction studies are established using surface electrodes. Segments covering the axilla, elbow, wrist, knee and ankle being especially studied because these areas include entrapment sites and are therefore territories in which conduction blocks are most likely to be found. A nerve biopsy in search of tomacula on nerve cross-sections or on teased fibres is sometimes performed, but because of its invasive nature it is not routinely used in the work up of HNPP.
Today genetic investigations and studies confirm the diagnosis when suspected by the above and promise to replace part of this process. As is discussed later, the discovery of a deletion on chromosome 17 that is causative of HNPP, and the ready availability of reliable and precise genetic tests offers a potential for a rapid and cheap confirmation of the diagnosis.
2.4.1 Differential diagnosis of HNPP and classification
The differential diagnosis of HNPP includes a number of diseases, mostly within the hereditary neuropathies (Meier and Moll, 1982; Pouget et al., 1992). Among these is a form of mononeuritis multiplex, namely hereditary mononeuritis multiplex with brachial predilection, or hereditary neuralgic amyotrophy. This disorder has an autosomal dominant inheritance and usually occurs in females where it is often associated with pregnancy. It is clinically and genetically distinct (Gouider et al., 1994; Chance et al., 1994; Pellegrino et al., 1996). In contrast to HNPP, it is painful and leads to amyotrophy and sensory loss affecting mainly the brachial plexus. However, both motor and sensory conduction velocities are normal (Windebank, 1994; Meier and Moll, 1982). Certain recurrent mononeuropathies associated with vasculitis in collagen diseases or diabetes mellitus should also be considered. Electrophysiology can show slowing of motor and sensory conduction velocities and therefore specific laboratory tests and typical nerve biopsy findings are required for them to be recognised. In some cases Charcot-Marie-Tooth 1 (CMT 1, HMSN-I, Peroneal muscle atrophy) should be considered in the differential diagnosis. This disorder, in which motor deficit predominates, results from a genetic abnormality in the same area of chromosome 17 as HNPP, a duplication rather than a deletion of the PMP 22 gene (see 3.0 Genetics and molecular understanding of HNPP). Histologically CMT 1 presents onion bulb formations of the myelin sheath that are different to the tomacula found in HNPP (see summary in Table I).
Combining previous clinical and electrophysiological observations with recent advances in the understanding of the molecular basis of diseases of the peripheral nervous system, several classification systems have been proposed. Schematically, there are two types of hereditary peripheral neuropathies: those affecting the axon and those affecting the Schwann cells of the myelin sheath. CMT 1 or hereditary motor and sensory neuropathy I (HMSN I), HNPP and Dejerine-Sottas neuropathy (DSS, HMNS III) are caused by abnormalities in Schwann cells and peripheral myelin (Lupski et al., 1997). CMT 2 (HMSN II), which can be distinguished electrophysiologically, appears to be caused by an axonal or neuronal defect (Murakami et al., 1996). Furthermore CMT 1 is sub classified genetically as either CMT 1A (autosomal dominant, usually linked to 17p11.2-p12 markers), CMT 1B (rare, linkage to 1q21.2-q23, or CMT 1C (autosomal dominant not linked to either chromosome 1 or 17) (Murakami et al., 1996). The other CMTs are also sub classified with regard to their specific genetic defects. X-linked CMT (CMT X) is associated with multiple mutations in the connexin 32 gene in the Xq13.1 region (Murakami et al, 1996). In males it presents similarly to CMT 1, whereas in females it shares certain electrophysiological traits with CMT 2.
| Tabl. I Disorders to be considered in the differential diagnosis of HNPP. Findings of HNPP are listed for comparison. |
| Disorder |
Clinical Presentation |
Inheritance
Pattern |
Age of
Onset |
Electro-physiological
Findings |
Histology |
Genetic
Defect |
| Hereditary neuropathy with liability to pressure palsies (HNPP) |
Chronic and recurrent peripheral palsies with paraesthesiae, particularly ulnar and peroneal nerves |
Autosomal dominant |
Predominantly 2nd-3rd decades |
Slowing of nerve conduction, conduction blocks at entrapment sites |
Tomaculous change in myelin sheath |
17p11.2 (PMP22) deletion, PMP22 point mutations |
| Hereditary mononeuropathy multiplex with brachial predilection |
One or several peripheral nerves involved, neuralgic amyotrophy, sensory loss |
Autosomal dominant |
|
Normal nerve conduction velocities |
|
|
| Charcot-Marie-Tooth 1 (CMT 1A, HMSN I) |
Pes cavus, congenital hip problems, motor deficit predominates |
Autosomal dominant |
2nd-3rd decades |
Slowing of nerve conduction |
Hypertrophic changes with onion bulb formations |
17p11.2 (PMP22) duplication, or point mutations |
| Mononeuropathies associated with diabetes, polyarteritis nodosa |
|
None |
Variable |
Nerve conduction can show slowings |
|
None |
| Déjerine - Sottas neuropathy (HMNS III) |
Severe limb weakness |
Autosomal recessive |
1st decade |
Slowing of nerve conduction |
Onion bulb formations |
Homozygous PMP22 duplication, point mutations |
3. Genetics and molecular understanding of HNPP
It has been known since the earliest descriptions of HNPP that the disease is transmitted in an autosomal dominant manner. In part due to its variable phenotype expressivity, the exact prevalence of HNPP is still unknown (Murakami et al., 1996). As an overall group of diseases, CMT is the most common inherited disorder of the peripheral nervous system. The prevalence of all types of HMSN is estimated at 1 in 10000 (Nelis et al., 1996). Authors propose that the frequency of the most common genetic defect associated with HNPP may be similar to that of the most common defect associated with CMT1 (Murakami et al., 1996) (see below). The most recent step towards understanding HNPP has come about as a result of genetic research involving the identification of a deletion on the short arm of chromosome 17. The deletion covers a 1.5 megabase region that includes the gene for peripheral myelin protein 22 (PMP22) (Matsumi et al., 1992; Chance et al., 1993; Mariman et al., 1993). The locus for this gene was first implicated by linkage analysis of affected families, in particular in the study of three pedigrees by Chance et al. (1993), but point mutations leading to frameshifts have since been discovered in patients, confirming its involvement (Nicholson et al.; 1994, Young et al., 1997; Bissar-Tadmouri et al., 2000). Clinical and genetic studies have recently showed that the 17p11.2 deletion is present in approximately 85% of patients with typical nerve conduction abnormalities or clinical presentations (Gouider et al., 1995; Tyson et al., 1996; Nelis et al., 1996). Linkage analysis and gene dosage studies provide some evidence of genetic heterogeneity underlying HNPP and other genes might be implicated (Mariman et al., 1994). Phenotypic heterogeneity in patients with different genetic mutations has also been described (Pareyson et al., 1996; Lenssen et al., 1998).
The PMP22 gene was first shown to be contained within the CMT 1A duplication (Timmerman et al., 1992). The exact role of the protein PMP22 is not known. It is an integral membrane glycoprotein. Studies have found it to be present principally but not exclusively in compact myelin of the peripheral nervous system. It is produced almost exclusively by Schwann cells (Snipes et al., 1992; Suter et al., 1993; Stögbauer et al., 2000). These authors suggest that it not only is a structural protein, but that it may also act as a channel or non-specific pore protein. The abnormal expression of the mRNA and the PMP22 protein has been proposed as the main determining factor in the molecular pathogenesis of a series of diseases including CMT 1A, DSS/HMNS III and HNPP (Yoshikawa et al., 1994; Schenone et al., 1997). It acts in a dose-dependent manner. Increased gene dosage leads to CMT 1A, whereas decreased gene dosage is found in HNPP (Stögbauer et al., 2000). In their study of mice deficient in the PMP22 gene (PMP220/0 mice), Adlkofer et al. (1995), concluded that PMP22 is required for the correct development of peripheral nerves, the maintenance of axons and the determination of myelin thickness and stability. Studies on PMP0/0 mice or on mice carrying the trembler (Tr) mutation in the PMP22 gene (Suter et al., 1992), systematically show walking difficulties as a consequence of progressive paralysis (Suter et al., 1993, Adlkofer et al., 1995). Other studies have confirmed that the PMP22 protein and its correct expression are essential for normal myelination (Trapp et al., 1996; Murakami et al., 1996) and normal Schwann cell growth (Suter et al., 1993).
The gene region that is deleted in HNPP is duplicated in CMT 1A (Valentijn et al., 1992), and is the first example in humans of Mendelian syndromes resulting from the reciprocal products of unequal exchange involving intra-chromosomal segments, particularly unequal crossover during meiosis (Chance et al., 1994). It provides the basis for furthering the understanding of meiotic recombination in humans (Reiter et al., 1998). Remarkably, the result of the duplication is, to some extent, similar to that of the deletion since CMT is part of the differential diagnosis (Roth and Magistris, 1984). Other mechanisms that can lead to the deletion have been proposed. Le Guern et al. (1996) have suggested that the deletion can be the result of intrachromosomal rearrangement in de novo cases of maternal origin and that this mechanism may be specific to maternal transmission.
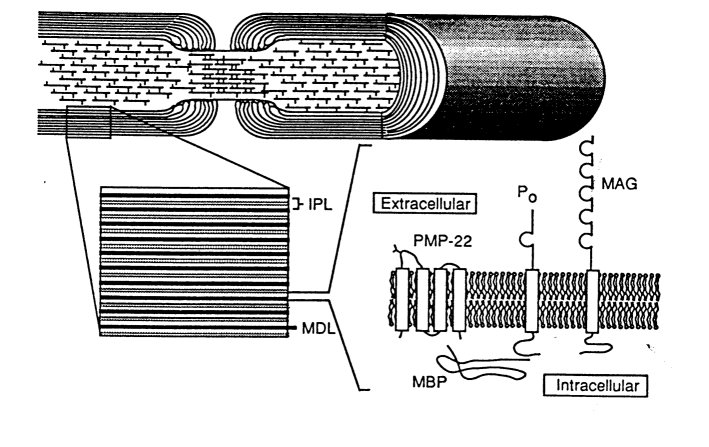 Fig. 4 Diagram showing relationship of proteins of myelin, including PMP22, and structure of the myelinated axon in the peripheral nervous system (MAG: myelin associated glycoprotein; MBP: myelin basic proteins; MDL: major dense line; IPL: intraperiod line; PMP22: peripheral myelin protein 22; P0: protein zero).
Reproduced from Suter et al., 1993.
Fig. 4 Diagram showing relationship of proteins of myelin, including PMP22, and structure of the myelinated axon in the peripheral nervous system (MAG: myelin associated glycoprotein; MBP: myelin basic proteins; MDL: major dense line; IPL: intraperiod line; PMP22: peripheral myelin protein 22; P0: protein zero).
Reproduced from Suter et al., 1993.
4. The investigation
4.1 Aim of the investigation
In light of a possible heterogeneity of presentation of HNPP patients, this research aims to determine whether all patients currently being followed for symptoms and complaints consistent with HNPP and having electrophysiological studies suggestive of HNPP, actually have the implicated deletion. The underlying question is therefore whether or not the clinical heterogeneity of HNPP is in fact a reflection of heterogeneous genetic defects. The potential benefits of the study include the advances it may provide with respect to our understanding of HNPP. The comparison of diagnoses made on classical criteria with those made on the basis of genetic investigations should give an indication of the efficiency and sensitivity of genetic diagnosis. The latter should prove valuable not only for symptomatic patients, but also for the diagnosis of asymptomatic family members. This development will allow better genetic counselling and preventive measures to be taken.
4.2 Methods
4.2.1 Study subjects and HNPP probability scoring
The individuals enrolled for this study were those that are currently being followed for, or had already been recognised, by the ENMG unit of the HUG Neurology Clinic as probably having HNPP based on clinical and electrophysiological findings. Twenty-four individuals from 22 families were studied. The investigation protocol was accepted by the University of Geneva Hospital ethics committee (Cométhique) on the 21 June 1995. All individuals included were investigated before 1997.
The nature of the research required minimal patient involvement. One blood sample of 9 ml per patient was taken. An electroneuromyographic control was carried out on patients who had not undergone a thorough examination beforehand or who had an ambiguous clinical picture. All individuals enrolled in the study were volunteers and, when desired, received genetic counseling. Special emphasis was given to possible preventive measures. Where it was indicated surgery was suggested.
After the initial enrolment, the individuals' clinical and electrophysiological findings were used to determine their respective probability of having HNPP. Four areas of particular value were tentatively used for the proposed clinical scoring system, namely personal and family history, clinical findings, electrophysiological findings. Nerve biopsy was done in only one patient and was therefore not used in the overall scoring. The relevance of each of these 4 categories was assessed for every individual and a score assigned to them. The total score was used to establish three overall groups of probability. Each individual was therefore placed into a group with either a low, moderate or high probability of having HNPP. This probability was then compared with the genetic results, the latter indicating whether or not a 17p11.2 deletion was detected. It might be noted that for some individuals not all the relevant information was available. In other instances certain features of the work up were deemed to be overwhelming and a presumptive diagnosis was made on, for example, the basis of the strong electrophysiological data. These subjects are marked with an asterisk (*) in table III.
Proposed scoring:
| Category |
Scoring for each category (indication of HNPP) |
Overall probability for each subject |
| Personal history |
0 = atypical or not done |
�6 = low |
| Family history |
1 = low |
7 - 9 = moderate |
| Clinical findings |
2 = moderate |
�10 = high |
| Electrophysiological findings |
3 = high |
|
4.2.2 Overview of the techniques
The main diagnostic technique used to detect the 1.5 Kb 17p11.2 deletion on chromosome 17 was fluorescent in situ hybridisation (FISH). This is a method whereby lymphocytes from heparinized blood are cultured in vitro. The lymphocytes are then fixed and transferred to slides. Cloned PMP22 gene probes are used to locate or confirm the absence of the concerned gene. The probe is associated with a fluorescent marker that enables direct microscopic analysis. This is the technique that was used in Geneva under the supervision of Dr Michael Morris (Laboratoire de Diagnostic Moléculaire, Division de Génétique Médicale, HUG). Twenty-one blood samples were tested using this method. Initially, and whilst the FISH was being developed in Geneva, some samples were tested using quantitative Southern blotting. In this technique, genomic DNA is cut with restriction enzymes and the resultant fragments are separated by size on a gel. After transfer onto a filter, a specific probe consisting of cloned cDNA is used to locate the desired gene or DNA segment. The gene copy number is estimated by quantification of the autoradiograph. This technique was used on 7 blood samples by Professor J. M. Burgunder (Laboratoire de Neuromorphologie, Clinique et Policlinique de Neurologie, Inselspital de Bern).
4.2.3 An introduction to fluorescent in situ hybridisation (FISH)
In situ hybridization can be used with several forms of genetic substrate, namely repetitive, single copy or cosmid probes and chromosome-specific libraries. It allows specific nucleic acid sequences to be detected in morphologically preserved chromosomes or in interphase nuclei. The technique was developed by Pardue and Gall (1969) and by John et al. (1969). Because molecular cloning was not initially available, this type of hybridisation could only be applied to genomic sequences that could be obtained and isolated using conventional methods. The radio labeling of the probes had the inconveniences associated with the handling of radioactive material. Specific oligonucleotide probes are now available and non-radioactive markers are used. Two nonradioactive hybridization methods can be distinguished, namely the direct and indirect procedures. In the direct method the detectable molecule (reporter) is bound directly to the nucleic acid probe so that formed hybrids can be visualized microscopically immediately after hybridisation with the target nucleic acid. The technique used in this study was an indirect procedure in which the probe contains a reporter molecule introduced chemically or enzymatically that renders it detectable by affinity cytochemistry. The reporter molecule has to be accessible to antibodies so that the full accuracy of this method is preserved. To optimize the precision and reliability of the results a two-colour in situ hybridisation procedure was used. This required the use of two reporter molecules, digoxigenin (DIG) and biotin. Digoxigenin is isolated from digitalis plants, namely Digitalis purpurea and Digitalis lanata. The blossoms and leaves of these plants are the only natural source of Digoxigenin and therefore no binding of the anti-DIG antibodies occurs in other biological material. The digoxigenin probes were revealed with fluorescein or FITC (Fluoresceinisothiocyanate) labelled antibodies, giving a green signal. The other molecule used was biotin, a vitamin of the B complex. Although the biotin probes could be detected using anti-biotin antibodies, in this case avidin was used. Avidin is a glycoprotein found in egg white and is used because of its high binding capacity with biotin. Coupled with rhodamine, this complex gives a red signal. Before incorporation into the probes both digoxigenin and biotin were linked to uridine (or deoxyuridine) nucleotides at the number 5 position. Boehringer Mannheim Biochemica, 1996.
4.2.4 Summary of the FISH procedure used in the study
For the two colour in situ hybridisation procedure two probes consisting of DNA sequences of chromosome 17 (17p11.2) included in the 1.5 Mb deletion of the PMP22 gene were used. The probes were cl03B11 and cl32G8. They were labelled with the reporter molecules using nick translation, whereby digoxigenin and biotin are linked to uridine nucleotides at the number 5 position of the pyrimidine ring, and incorporated enzymatically into the DNA probe. The two enzymes used were DNase I, which is an endonuclease, and DNA polymerase I. As RNA inhibits the polymerase, RNase is used to eliminate any residual RNA. EDTA is added in order to chelate Mg2+ and to arrest the enzyme activity. Heparinized blood obtained from the subjects was cultured for three days in a RPMI 1640 culture medium with phytohemaglutinine and gentamycine, and then treated with colchicin to arrest lymphocytes in metaphase. After exposure to a hypotonic solution and fixation with a methanol/acetic acid solution, the suspensions were "dropped" onto clean slides, which were then briefly rinsed with acetic acid to remove residual cytoplasm. A "mixture" (Herring sperm DNA, yeast tRNA, human Cot-1 DNA, the labelled probe) then underwent pre-annealing or chromosomal in situ suppression (a process by which introns and non relevant probe DNA are hybridised prior to exposure to the sample to be tested so that they do not interfere with the specificity of the procedure; the Herring sperm and Cot-1 DNA bind with the these sequences). After the denaturing of the fixated chromosomes (done by immersing the slides in a formamide/SSC (sodium citrate solution)), the slides were placed in an ethanol solution to prevent reassociation, and let to dehydrate. The hybridisation mixture was then placed on the prepared slides. Post hybridization steps included several washes and detection of the hybridized digoxigenin and biotin probes with high affinity fluorescein (FITC) labelled anti-digoxigenin antibodies and rhodamine labelled avidin, respectively. FITC appears as green, and rhodamine appears as red. High power microscopic analysis was performed to visualise the chromosomes and to verify whether hybridization had occurred (Gos, 1993).
5. Results
5.1 Overview
The 17p11.2 deletion was found in 7 of the 24 individuals studied; six of these were found using the FISH technique and one with the Southern blotting. Where both diagnostic techniques were used (4 cases), the results correlated perfectly. Of the individuals presenting with the deletion, 4 were highly suspected of having HNPP on the basis of the clinical and electrophysiological findings. The deletion was found in 2 patients in whom suspicion of HNPP was moderate, and in one whose findings suggested a low probability. In one family, of the 3 members investigated, 2 had the deletion.

5.2.1 Clinical and laboratory data
The following tables summarize the relevant information and findings for each individual that was included in this study. Table II displays the clinical information available, as well as the results of the genetic tests that were carried out. Table III displays the interpretation and systematic scoring of the different categories described in the previous chapter. Each individual was assigned to a group that indicates with what probability, low, moderate or high, HNPP was suspected. In some cases an individual was placed into a high probability group, even though all information was not available; in other instances certain features of the work up were deemed to be overwhelming and a presumptive diagnosis was made on, for example, the basis of the strong electrophysiological data. These cases are marked with an asterisk (*) in table III. Figure 6 summarizes the number of individuals in each probability category found to have a deletion.
| Tabl. II Summary of the clinical and electrophysiological findings and genetic results |
| Subject |
Year of birth, age at time of testing |
Sex |
Clinical findings and results of biopsies if carried out |
Electrophysiological findings and or conclusions |
Family History |
Genetics |
| |
|
|
|
FISH |
Sb |
| 1 |
1933,
62 years |
F |
One-month history of weakness in right hand following right upper limb immobilisation after surgery for rotator cuff tear. No amyotrophy. Important hypothenar weakness. Hypoesthesia in fingers. |
Right ulnar nerve conduction block at elbow. Sensory impairement of right radial nerve (low size potential, slowing of conduction velocity). Carpal tunnel syndrome. |
Incomplete family history. |
Del |
Not
done |
| 2 |
1972,
23 years |
M |
Following ski accident, 1-month history of parasthesiae on ulnar side and two last fingers of right hand. History of repeated spasmodic torticollis. |
Right ulnar nerve conduction block at elbow. Sensory and motor fibre involvement predominantly 15 mm above medial epicondyle. |
No relevant family history. |
No Del |
No Del |
| 3 |
1947,
28 years |
F |
Motor and sensory deficit in left ulnar nerve after compression at the elbow. Status post bilateral carpal tunnel syndrome. 2-3 history of burning type sensation in right thigh area. |
Left ulnar nerve 60% conduction block at elbow in sulcus nervi ulnaris, focal slowing of conduction velocity. Several affected nerves with conduction blocks at entrapment sites. |
No clear family notion of neurological problems. |
No Del |
Not
done |
| 4 |
1939,
57 years |
F |
Patient investigated for right foot drop, evolving over several years with progressive worsening. Bilateral carpal tunnel syndrome. Sensory and motor deficits in right lower limb, bilateral achillean areflexia. |
Sensory and motor deficit of peripheral nervous system. Mainly axonal impairment, few signs of acute denervation, some signs of chronic de- and reinervation (large motor unit potentials). No predominance of affection at entrapment sites. |
Incomplete family history |
No Del |
Notdone |
| 5 |
1979,
17 years |
M |
Right foot drop, which appeared after a hike, with no other apparent cause. No associated pain. First episode. Total deficit of muscles of anterior compartment of lower leg, hypoesthesia on dorsum of foot. |
Mixed affection of common peroneal nerve at the neck of fibula, with 50% axonotmesis and 50% neurapraxia. |
No family history of neurological problems. |
No Del |
Not done |
| 6 |
1917,
78 years |
F |
Decreased proximal strength in lower limbs with history of fatigability. History on non-insulin-dependant diabetes mellitus. History of numbness and paraesthesia in feet and to a lesser extent in 4 and 5 right fingers. Autonomic disturbances. Muscle biopsy: slight muscular damage, compatible with diabetic neuropathy. |
Sensory, motor and autonomic polyneuropathy prevailing in lower limbs. Moderate slowing of conduction velocity of median nerves at carpal tunnel bilaterally and of right ulnar nerve at wrist. No focal slowing, no conduction blocks. |
No relevant family history. |
No Del |
Not done |
| 7 |
1990,
6 years |
F |
Young girl with scoliosis and areflexia in all four limbs. |
Diffuse myelinic type motor neuropathy, possibly CMT1. |
No family history of neuromuscular disorders. |
No Del |
Not done |
| 8 |
1928,
67 years |
M |
Neuropathy with HNPP type characteristics. Nerve biopsy done. |
Multifocal conduction blocks. |
No family history available. |
Del |
Not done |
| 9 |
1952,
42 years |
M |
6 week history of pain in right external sub-clavicular region and right shoulder blade area, radiating to thumb, accompanied by dysesthesia. Moderate right triceps amyotrophy and weakness. History of numbness in upper limbs after wind surfing. History of rapid onset and painless proximal left upper limb paralysis 7 years earlier, with improvement over a 2 months period. Bilateral carpal tunnel syndrome. |
At time of prior upper limb paralysis, conduction block found on axillary nerve as well as confirmation of bilateral carpal tunnel syndrome. No conduction blocks at latest examination. |
No known neurological problem in parents. Sister operated for carpal tunnel syndrome. |
No Del |
Not done |
| 10 |
1955,
39 years |
M |
One-month history of weakness in right foot extensor muscles. No amyotrophy. Discrete sensory deficits on dorsum of foot. |
Principally a neurapraxic type impairment of right common peroneal nerve, with 85% conduction block at the neck of the fibula. No abnormality of ulnar nerve at elbow. |
No family history of neuromuscular problems. |
Not done |
No Del |
| 11 |
1923,
61 years |
M |
Four year history of left L4 sciatica with lower limb hyperreflexia and right foot extensor paresis (narrow lumbar canal on MRI). Two-year history of bilateral ulnar nerve territory hypoesthesia and paresthesia in fingers. Left ulnar nerve appears thickened on palpation. Left bicipital and right tricipital hyporeflexia. |
Multineuropathy with chronic and "ancient" signs. No polyneuropathy. Bilateral slowing of ulnar nerve conduction velocity predominating in sensory fibres. No conduction abnormality at elbow. Signs of acute and chronic denervation in lower limbs. No conduction blocks. |
No HNPP type history in parents or in the rest of the family. |
Not done |
Del |
| 12 |
1993,
17 months |
F |
Child treated with harness for left hip dislocation between 2 and 6 months of age. Symmetrical bilateral lower limb flaccid paralysis observed 2 weeks after end of treatment, with seemingly intact sensory function. |
Motor neurography showing very reduced evoked potential amplitude. Normal sensory neurography. Indicative of peripheral nerve impairment which only concerns motor fibres. |
Incomplete family history. |
No Del |
Not done |
| 13 |
1961,
34 years |
F |
Two months history of left hand sensory deficit and clumsiness, noticed on waking up one morning. Clinically, moderate motor and sensory deficits in left ulnar nerve territory. History of left common peroneal nerve affection, with rapid complete recovery. |
65% conduction block of left ulnar nerve localized at elbow in groove for ulnar nerve, with slowing of conduction velocity. Normal sensory function of median, radial and ulnar nerves. In 1981, left common peroneal nerve conduction block demonstrated at neck of fibula. |
Incomplete family history. |
No Del |
Not done |
14
family A |
1945,
49 years |
M |
Several weeks' history of left thenar area hypoesthesia; occurred after carrying shopping bag (compression by bag handle). History of many similar events, all regressing after some time. Cannot remain seated in same position for extended period of time without having paraesthesia and cramps. Repeated paraesthesia and cold sensation in hand since childhood. Carpal tunnel syndrome surgery in 1987, at age 42. |
Predominantly myelinic sensory and motor polyneuropathy with slowing of conduction velocities at entrapment sites. Motor neurography shows low amplitude evoked potentials. Conduction block at right elbow. Prior exam showed median nerve conduction block between wrist and palm. |
Many similar episodes in son (N°17). Brother undergone neurosurgical interventions. Sister hospitalised with a "myopathy". Father had left lower limb "neurological problem" Uncle hospitalised for paralysis following discal hernia surgery. |
Del |
Del |
15
family A |
1971,
24 years |
F |
Sister of N°15. No complaints or history of neurological problems. Slight tendency to have paraesthesiae or certain sensations in lower limbs if remains in same position too long, for example at the cinema. Raynaud's phenomenon has been evoked. Normal strength and no sensory problems. |
Electrophysiological results within normal limits. |
Strong family history of neurological problems. For brother see N°17, for father see N°15. Uncle and great uncle had neurological problems. |
No Del |
No Del |
16
family A |
1967,
26 years |
M |
Son of N°15. Two-month history of left foot drop, with no known triggering factor or event. Sensory problems on antero-external part of lower leg and foot. Had significant weight loss (10 Kg). This is first significant episode but has had previous trouble with maintaining certain positions such as crossed legs. |
Myelinic predominant sensory and motor polyneuropathy. Common peroneal nerve conduction block at the neck of fibula. |
Strong family history of neurological problems. Father has had many similar episodes (see N°15). Uncle and great uncle have had neurological problems. |
Del |
Del |
| 17 |
1960,
35 years |
F |
Neuropathy with HNPP type characteristics. Incomplete information |
Incomplete information |
Incomplete information. |
No Del |
Not done |
| 18 |
1945,
49 years |
M |
Several month history of sensory problems in hands on waking, with complaints of paresthesia and numbness (started in left hand). Pain in forearm musculature when exercising. Occasionally similar symptoms in toes. |
Sensory and motor conduction abnormalities of median nerves, mainly distally suggesting myelinic type impairment. No conduction blocks detected on tested nerves. No signs of reinnervation (no MARs ). |
Family history of rhumatology problems (arthritis in sisters). No history of neurological problems. |
Not done |
No Del |
| 19 |
1970,
16 years |
F |
Pain in upper right shoulder area. Status post surgery for shoulder instability following car accident in 1992 (5 years prior to present exam). Three-month history of decreased sensation in right thumb. History of frequent distal hypoesthesia and paraesthesia in right hand on waking. |
Moderate bilateral carpal tunnel syndrome (subnormal bilateral motor latencies and subnormal right sensory conduction velocity). |
Incomplete family history. |
No Del |
Not done |
| 20 |
1928,
71 years |
F |
Right foot drop since 1963, following delivery. Fluctuating bilateral sensory and motor problems. Patient has noticed fasciculations in certain muscles. Neurolysis of right median nerve at wrist and right ulnar nerve at elbow in 1994 with good evolution.Nerve biopsies show tomacula. |
Myelinic and axonal sensory and motor polyneuropathy with conduction blocks. Right ulnar nerve conduction block at elbow > ; 50%. Sensory neurography of right ulnar and median nerves shows small amplitude responses. |
Incomplete family history. |
Del |
Not done |
| 21 |
1923,
72 years |
M |
Chronic polyneuropathy suggestive of CMT1 or HNPP. |
Incomplete information. |
Incomplete family history. |
No Del |
Not done |
| 22 |
1941,
53 years |
F |
History of recurrent truncular nerve impairments. Ulnar nerve impairment following unusual type of work. |
Diffuse slowing of nerve conduction velocities, but predominating at entrapment sites. Persistent ulnar nerve conduction block at elbow (following unusual work). |
Family history of similar recurrent nerve impairments. |
Del |
Not done |
| 23 |
1930,
64 years |
M |
Bilateral lumbar-sacral radiculo-plexopathy since 1989 with global weakness, though predominantly in left lower limb. Both lower limbs affected in 1994. Aggravation in 1995. |
Axonal type sensory and motor peripheral nerve impairment of lower limbs. Bilateral ulnar nerve conduction blocks at elbows with axonal type neuropathy. Slight bilateral slowing of distal median nerve conduction velocities. |
No relevant family history. |
No Del |
Not done |
| 24 |
1981,
15 years |
M |
Patient with cystic fibrosis. Symmetrical lower limb weakness following pulmonary transplant 9 months earlier. Diminished sensory function below knees. |
Lower limb results compatible with a severe axonal type sensory and motor neuropathy. |
Incomplete family history. |
No Del |
Not done |
| Tabl. III Probability of HNPP, according to proposed scoring (low �6, moderate 7-9, high �10). |
| Subjects |
Personal history |
Clinical findings |
Family history |
Electrophys-
iological findings |
Total
X/12 |
Probability |
Deletion |
| 1* |
2 |
2 |
0 |
3 |
7 |
High |
Yes |
| 2 |
2 |
1 |
0 |
3 |
6 |
Low |
No |
| 3 |
3 |
1 |
0 |
3 |
7 |
Moderate |
No |
| 4 |
2 |
2 |
0 |
3 |
7 |
Moderate |
No |
| 5* |
2 |
2 |
0 |
1 |
5 |
Low |
No |
| 6 |
2 |
1 |
0 |
2 |
5 |
Low |
No |
| 7 |
1 |
1 |
0 |
1 |
3 |
Low |
No |
| 8 |
2 |
2 |
0 |
3 |
7 |
Moderate |
Yes |
| 9 |
1 |
2 |
1 |
2 |
6 |
Low |
No |
| 10 |
3 |
2 |
0 |
3 |
8 |
Moderate |
No |
| 11 |
2 |
2 |
0 |
2 |
6 |
Low |
Yes |
| 12 |
1 |
2 |
0 |
1 |
4 |
Low |
No |
| 13 |
3 |
2 |
0 |
3 |
8 |
Moderate |
No |
| 14 |
3 |
3 |
3 |
3 |
12 |
High |
Yes |
| 15 |
2 |
1 |
3 |
0 |
6 |
Low |
No |
| 16 |
3 |
2 |
3 |
2 |
10 |
High |
Yes |
| 17* |
2 |
2 |
0 |
0 |
4 |
Low |
No |
| 18 |
3 |
2 |
0 |
1 |
6 |
Low |
No |
| 19 |
1 |
2 |
0 |
2 |
5 |
Low |
No |
| 20 |
3 |
3 |
0 |
3 |
9 |
Moderate |
Yes |
| 21* |
3 |
0 |
0 |
2 |
5 |
Low |
No |
| 22 |
3 |
2 |
3 |
3 |
11 |
High |
Yes |
| 23 |
2 |
2 |
0 |
3 |
7 |
Moderate |
No |
| 24 |
2 |
2 |
0 |
1 |
5 |
Low |
No |
| * Indicates individuals on whom there is incomplete information, but with certain important characteristics in given categories. |
 Fig. 5 Age distribution of the 24 individuals included in the study (age at time of genetic testing).
Fig. 5 Age distribution of the 24 individuals included in the study (age at time of genetic testing).
 Fig. 6 Graph showing the number of patients in each probability category and the number of patients in that category in whom the 17p11.2 deletion was detected.
Fig. 6 Graph showing the number of patients in each probability category and the number of patients in that category in whom the 17p11.2 deletion was detected.
5.2.2 Laboratory (FISH) data
The key findings obtained with the FISH are described below. The lymphocytes are blocked in metaphase. Figure 7 shows FISH results for individuals with no 17p11.2 deletion; both probes can be seen on both chromosomes 17. Figures 8 and 9 show positive results that are cases in which the analysed lymphocyte chromosomes present a deletion on chromosome 17. As labelled on the figures, two cosmids are visible: Cosmid 1 corresponds to the cl03B11/DIG probe and is revealed with the green fluorescein (FITC) coupled antibodies. Cosmid 2 corresponds to the cl32G8/biotin probe and is revealed with the red avidin/rhodamine complex. Each probe generates two signals per chromosome, one on each chromatid. In figures 8 and 9 the two visible chromosomes 17 show different patterns: on one chromosome 17, both cosmids are visible, on the other neither cosmid can be seen, the absence of hybridised probes demonstrating that there is a deletion. In some pictures a third marker is present on each chromosome 17. This marker corresponds to a probe that binds to the tip of chromosome 17 in an area not concerned by the typical deletion. It is used as a control to indicate that hybridisation has occurred and facilitates the identification of the chromosomes 17. Without this marker, conventional morphological traits are necessary to identify the chromosomes. The fact that chromosomes are being blocked in metaphase of mitosis means that both chromatids are not yet separated. Figure 8 is the result obtained for subject 9 and figure 9 that for subject 1.
FISH can also be used to detect the CMT 1A type 17p11.2 duplication. An example of this duplication is shown in figure 10. In this case there are three pairs of PMP22 probes, but only two control probes, indicating that one homologous chromosome 17 has a duplicated region. In this case the lymphocyte nucleus is blocked in interphase.
 Fig. 7 FISH result of a control case showing no 17p11.2 deletion. Both PMP22 probes are visible on both chromosomes 17. Note the presence of the control probe on both chromosomes.
Fig. 7 FISH result of a control case showing no 17p11.2 deletion. Both PMP22 probes are visible on both chromosomes 17. Note the presence of the control probe on both chromosomes.
 Fig. 8 FISH analysis of patient 9 showing a hemizygous deletion on one of the chromosomes 17. Both PMP22 probes are visible on one of the chromosomes but are absent on the other
Fig. 8 FISH analysis of patient 9 showing a hemizygous deletion on one of the chromosomes 17. Both PMP22 probes are visible on one of the chromosomes but are absent on the other
 Fig. 9 FISH result of subject 1. Hemizygous deletion. In this instance no control probe was used to identify the chromosomes. Arrows identify chromosomes 17.
Fig. 9 FISH result of subject 1. Hemizygous deletion. In this instance no control probe was used to identify the chromosomes. Arrows identify chromosomes 17.
 Fig. 10 Fish analysis of a patient with CMT 1a. There are three pairs of PMP22 probes but only two control probes. The lymphocyte nucleus is blocked in interphase
Fig. 10 Fish analysis of a patient with CMT 1a. There are three pairs of PMP22 probes but only two control probes. The lymphocyte nucleus is blocked in interphase
6. Discussion
Individuals presenting with: a) an autosomal dominant neuropathy, b) chronic and recurrent complaints of palsies and parasthesiae which can be put into relation to minor trauma, and c) in whom electrophysiological studies show nerve conduction velocity slowing and conduction block, have a strong likelihood of having HNPP. A 1.5 Mb deletion on chromosome 17p11.2 is considered specific to HNPP and therefore serves as a definite confirmation of the diagnosis. The deletion may also be found in asymptomatic individuals and family members, and molecular genetic analysis is of value for detecting at-risk individuals.
Twenty-four individuals with suspected HNPP were included in this study. The 17p11.2 deletion was found in 7 of the 24 individuals studied. The probability score used was shown to give reasonably accurate predictions of HNPP diagnosis; all four individuals who were in the high probability group had the 17p11.2 deletion. It is notable that deletions were detected in two individuals in whom the suspicion was moderate, and in one whose suspicion of HNPP was low. Personal and family history, clinical and electrophysiological findings, taken individually, were poorly correlated with the genetic results. However, taken as a composite score there was a better correlation. Based on electrophysiological findings only, of the 7 individuals found to have a deletion, 5 had a high, and 2 had a moderate probability. A deletion was not found in 6 cases where the electrophysiological examination showed typical signs. This indicates that although the value of genetic testing is clear it is also true that some conventional findings are sensitive parameters in the diagnostic process. In some instances only genetic testing can formally exclude the disease. For example, in instances where some members of a family are affected, definite results can be very useful in identifying asymptomatic family members. Genetic results can be used to reassure the individual or, if positive, to provide counselling on preventive measures to be taken. Only genetic testing is of true value in presymptomatic patients, and is obtained by a single test for life. The comparison of pre-test scoring and genetic results on a greater number of subjects, including asymtomatic family members, in a double blinded protocol could provide useful information for the elaboration of a more accurate scoring tool, or to determine subgroups of individuals in whom genetic testing might be particularly useful.
This study supports, on a small scale, what other authors (Gouider et al., 1995; Tyson et al., 1996) have described, namely that the 17p11.2 deletion is the principal genetic defect found in HNPP. FISH provides a precise method for detecting specific defects but it can only detect abnormalities that are defined by the particular sequences coded by the chosen probes. In this study the fact that the deletion was not found in all potential cases suggests the possibility of smaller defects such as point mutations, examples of which can be found in the literature (Nicholson et al., 1994; Young et al., 1997; Bissar-Tadmouri et al., 2000).
The precise aetiology of HNPP remains unknown. The disorder is thought to result from the abnormal expression of a gene coding for the PMP22 protein, which is normally found in compact myelin. The under expression of this protein may affect not only the structure of myelin but the actual function of Schwann cells (Suter et al., 1993; Schenone et al., 1997). The fact that in individuals presenting with the 1.5 Mb deletion on chromosome 17, an intact copy of the segment exists on the homologous chromosome might also explain some of the heterogeneity of the disorder. A dose response mechanism where by the quantity of PMP22 produced is the determining factor in the clinical presentation has been suggested. A functional polymorphism of the remaining copy of the gene could also lead to heterogeneity. PMP22 is an integral membrane glycoprotein and it is therefore conceivable that mutations in the gene lead to either the total absence of the protein or to an abnormal function. Some authors have suggested that some heterozygous mutations might lead to the formation of mutant proteins that hinders the proper function of the remaining half normal PMP22 protein, or alternatively that certain transmembrane domains are altered (Pareyson et al., 1997). Insights into PMP22 protein function might be obtained from precise phenotype-genotype comparisons of specific mutations.
The limitations of FISH probably mean that, although this technique is efficient for detecting the deletion, it is mainly useful in confirming the diagnosis of HNPP. When no deletion is detected, the possibility - however slight - of point mutations or other defects cannot be excluded. Carrying out direct sequence analysis is perhaps the only way to completely understand the genetic defects found in individuals with HNPP. However, according to present literature and the overwhelming importance of the 1.5 Mb deletion in most affected individuals (Nelis et al., 1996), as well as the relatively high cost, this is probably not justified in a routine clinical workup.
In spite of its limitations, FISH presents many advantages. It is, comparatively, a simple technique, which allows for rapid results and is relatively inexpensive, making it a useful and potent clinical tool.
Hereditary neuropathy with liability to pressure palsies is a disorder that has been known to exist for fifty years. It is a condition with a variable clinical expression. Although there is no curative treatment for this neuropathy, being able to provide patients with a diagnosis is often in itself an important step. The condition is one in which simple preventive measures to avoid nerve trauma can greatly reduce the potential disability associated with HNPP.
With the advent of modern molecular analysis, rapid and effective diagnosis is becoming a reality. Although a thorough history, physical exam and complete electrophysiological exam still form the basis of the diagnosis, genetic testing can now offer a significant aid. It is of particular interest in patients with an atypical clinical and electrophysiological presentation. Confirming the presence of the genetic defect has implications for the individual as well as for other family members. Genetic counselling is an essential part of the care and management of patients. FISH provides an efficient way of obtaining ready confirmation of the diagnosis, and therefore has the potential to become a valid clinical tool. It is important to understand that FISH is useful principally when it provides a positive result; when negative results are obtained, possible point mutations or other disorders affecting the expression of PMP22 cannot be excluded.
One of the aims of this study was to verify whether or not the diverse clinical presentation of HNPP is a reflection of a genetic heterogeneity. Although the results do not provide a conclusive answer, the fact that, in this study, the deletion was not detected in all patients who had a high probability of having the disorder, lays the groundwork for further investigations to be carried out. In particular direct sequence analysis could provide interesting insights into the disease and its presentation.
[Previous]
[Next]