2. Les méthodes de calculs utilisés
2.1 L'équation de Schrödinger
2.1.1 Formulation générale
Toute l'information que l'on peut obtenir sur un système constitué d'un ensemble de particules est contenue dans la fonction d'onde Y du système. La fonction d'onde d'un système composé de N atomes et 2n électrons est obtenue en résolvant l'équation de Schrödinger indépendante du temps suivante [1] :
HY = EY (1)
où E est l'énergie du système et H est l'opérateur correspondant : l'hamiltonien du système. Y est la fonction d'onde du système, fonction des coordonnées des noyaux, des électrons et contient toute l'information du système, E est l'énergie totale. Les valeurs propres de H sont les valeurs observables de cette énergie et les fonctions d'onde correspondantes sont les fonctions propres associées.
Comme il l'a été dit au chapitre I, les propriétés moléculaires qui peuvent être calculées par la résolution de l'équation de Schrödinger sont la géométrie moléculaire, et donc les stabilités relatives, les spectres de vibrations, les moments dipolaires et quadripolaires, les spectres électroniques et aussi des fonctions descriptives de la réactivité, telles que les charges atomiques et les fonctions de Fukui. Toutefois, la précision avec laquelle on peut espérer calculer ces quantités est très variable en fonction de la nature de ces propriétés. Cette équation ne peut en effet pas être résolue de manière exacte pour les systèmes moléculaires, et l'on doit donc effectuer un certain nombre d'approximations.
Pour un système traité comme étant composé de charges ponctuelles (2n électrons et N noyaux), sans traitement relativiste, l'hamiltonien pour un système à couches fermées est donné par :

 est la constante de Planck h divisée par 2p, me est la masse de l'électron, e est la charge de l'électron, MA est la masse du noyau A, rkA est la distance entre l'électron k et le noyau A, RAB est la distance entre les noyaux de l'atome A et de l'atome B dont les charges nucléaires sont respectivement ZA et ZB.
est la constante de Planck h divisée par 2p, me est la masse de l'électron, e est la charge de l'électron, MA est la masse du noyau A, rkA est la distance entre l'électron k et le noyau A, RAB est la distance entre les noyaux de l'atome A et de l'atome B dont les charges nucléaires sont respectivement ZA et ZB.  est le laplacien du kième électron défini de la manière suivante :
est le laplacien du kième électron défini de la manière suivante :

Cet hamiltonien ne prend pas en considération les interactions entre les électrons et des champs extérieurs au système (par exemple RPE) ou entre les électrons et les spins nucléaires (par exemple RMN) ; elle est indépendante du temps.
On constate que l'équation de Schrödinger, basée sur cet hamiltonien, est difficilement applicable à des molécules polyatomiques ; on doit donc introduire des approximations telles que l'approximation de Born-Oppenheimer et l'approximation orbitale pour la résoudre.
On utilisera par la suite les notations en unité atomiques. Dans ce système d'unités me = 1 ; = 1, e = 1 et 4peo = 1. On assumera d'autre part que le système étudié est à couches fermées.
Grâce à l'utilisation des unités atomiques, l'hamiltonien se simplifie sous la forme :

2.1.2 L'approximation de Born-Oppenheimer
En 1927, Born et Oppenheimer ont proposé de simplifier la résolution de l'équation (1) en séparant la partie électronique de la partie nucléaire dans la fonction d'onde Y. Cette approximation est basée sur le fait que les électrons se déplacent beaucoup plus rapidement que les noyaux, ceci étant dû à la masse beaucoup plus faible des électrons (environ 1836 fois moindre de celle du proton). Par conséquent, les électrons réagissent quasi instantanément à une modification de la position des noyaux [2].
En d'autres termes, pour une conformation R donnée des noyaux, seule la contribution électronique e(R) à l'énergie totale E est nécessaire pour connaître les propriétés du système. Cela revient donc à résoudre deux équations du type Schrödinger, l'une pour la partie nucléaire et l'autre pour la partie électronique. La fonction d'onde du système, solution de l'équation de Schrödinger dans l'approximation de Born et Oppenheimer, peut donc s'écrire sous la forme d'un produit de deux fonctions :

où F(R) est la fonction d'onde nucléaire, YR(r) est la fonction d'onde électronique correspondant à un jeu de positions R des noyaux figés, r et R étant respectivement les positions des électrons et des noyaux.
En écrivant l'hamiltonien H sous la forme :

où V(r,R) est un potentiel dépendant de la position des électrons et des noyaux, on fait apparaître un opérateur électronique He(r;R) de la forme :

On peut montrer, moyennant certaines approximations, que si l'on remplace l'expression (2) dans l'équation de Schrödinger, on obtient :

la fonction d'onde Ye(r) est une fonction propre de l'opérateur électronique He avec la valeur propre e(R), pour des positions R des noyaux figées. En résolvant l'équation (3) pour plusieurs positions successives des noyaux, on obtient alors une fonction de R :

qui représente l'énergie Born-Oppenheimer du système en fonction des positions R des noyaux immobiles.
Born et Oppenheimer ont aussi montré que le mouvement des atomes est régi par une équation de type Schrödinger où le potentiel dépend de l'énergie électronique évaluée par l'équation (3) :

U(R) joue donc le rôle d'une énergie potentielle pour le mouvement des noyaux. L'ensemble des conformations R des atomes permet alors de construire une surface d'énergie potentielle appelée « surface de Born-Oppenheimer (BO) ». Il s'agira d'une fonction à 3N-6 variables (3N-5 pour les molécules linaires) dont les minima correspondent aux géométries stables de la molécule. Au minimum de plus basse énergie correspond la géométrie à l'équilibre de la molécule. La détermination de U(R) et de ses dérivées première et seconde permet de localiser des points stationnaires sur la surface de BO et, par conséquent, d'élaborer des chemins réactionnels. Elle donne aussi accès aux constantes de force des molécules et donc aux fréquences de vibrations, de même que peuvent être calculées des propriétés telles que le moment dipolaire, la polarisabilité, etc.
Pour la résolution de la partie électronique, en considérant que le comportement des électrons n'est pratiquement pas modifié par les faibles déplacements des noyaux que l'on suppose comme étant figés dans leur position instantanée, l'hamiltonien dans l'approximation de Born-Oppenheimer se limite aux composantes électroniques seules :

On remarque cependant que le dernier terme  est un opérateur biélectronique alors que les deux premiers sont monoélectroniques, ce qui pose une difficulté ultérieure pour le traitement de la fonction Ye.
est un opérateur biélectronique alors que les deux premiers sont monoélectroniques, ce qui pose une difficulté ultérieure pour le traitement de la fonction Ye.
2.1.3 L'approximation orbitale
La fonction d'onde électronique Ye (que nous désignerons dorénavant uniquement par la lettre Y) est une fonction des coordonnées de tous les électrons du système. Si 2n est le nombre d'électrons (2n est choisi ici par comodité), Y est une fonction à (2n)×3 variables que l'on note communément Y(1,2,... 2n).
L'approximation orbitale, introduite par Hartree en 1928 [3], consiste à découpler les 2n électrons en développant la fonction Y(1,2,...,2n) en un produit de 2n fonctions monoélectroniques, de sorte que :

où l'indice i désigne l'orbitale i
Cette situation correspond physiquement à un modèle de particules indépendantes dans lequel chaque électron se déplace dans un champ moyen créé par les noyaux et la densité électronique moyenne des autres électrons. Cela signifie que chaque électron ressent les autres en moyenne, ce qui constitue naturellement une approximation.
La fonction d'onde n'a cependant pas de terme décrivant le spin car celui-ci est absent de l'hamiltonien électronique. Pour décrire complètement la distribution des électrons, la coordonnée de spin s doit donc être introduite, et celle-ci prendra les valeurs +1/2 ou -1/2. Le spin est une propriété intrinsèque de l'électron, de nature purement quantique, et n'a donc pas d'équivalent en mécanique classique. La fonction d'onde de spin pour le spin aligné le long de l'axe (+)z sera a(s) et celle pour le spin aligné le long de (-)z sera b(s).
La fonction d'onde électronique est donc composée d'une partie spatiale, l'orbitale, et d'une partie de spin. La fonction f est ce que l'on appelle une spin-orbitale et on l'écrit :
f(r,s) = c(r)h(s)
où r et s sont les coordonnées d'espace et de spin, respectivement.
Pour un système à 2n électrons la fonction d'onde polyélectronique Y la plus simple s'écrira donc sous la forme d'un produit de spin-orbitales supposées normalisées :

La fonction d'onde représentée par l'équation ci-dessus n'est cependant pas encore complète, car elle ne prend pas en compte l'indiscernabilité des électrons, ni le principe d'exlusion de Pauli [4]. Celui-ci a montré que pour les fermions (particules à spin ½), une spin-orbitale doit être antisymétrique par rapport à la permutation impaire des coordonnées d'espace et de spin. En permutant deux électrons il vient, par exemple :

Une telle fonction obéit au principe d'exclusion de Pauli qui impose à deux électrons de ne pas pouvoir occuper la même spin-orbitale, ainsi qu'à l'indiscernabilité des électrons. Or, dans la formulation de Hartree de la fonction d'onde, cela n'est pas le cas, car l'électron i occupe précisément la spin-orbitale i.
Hartree et Fock ont généralisé ce concept en montrant que le principe d'exclusion de Pauli est respecté si l'on écrit la fonction d'onde sous la forme d'un déterminant construit à partir de n spin-orbitales [5] ; on obtient alors ce qui est connu sous le nom de « déterminant de Slater » :

Les variables xi représentent ici les coordonnées d'espace et de spin.  est le facteur de normalisation ; 2n étant le nombre d'électrons.
est le facteur de normalisation ; 2n étant le nombre d'électrons.
On constate que la forme déterminantale de la fonction d'onde respecte le principe de Pauli : l'inversion de deux électrons correspond à la permutation de deux lignes (ou de deux colonnes), ce qui a pour effet de changer le signe du déterminant. Les spin-orbitales fi doivent, d'autre part, être différentes les unes des autres, car dans le cas contraire, le déterminant (6) s'annule.
Le problème consiste dés lors à rechercher les meilleures spin-orbitales conférant l'énergie la plus basse possible au système, conformément au principe variationnel ; ce but est atteint un utilisant la méthode auto-cohérente de Hartee-Fock.
2.1.4 La méthode de Hartree-Fock
Après avoir défini la forme de la fonction d'onde électronique globale d'un système polyélectronique à 2n électrons, il nous faut encore trouver l'expression de l'énergie électronique de ce système. D'autre part il nous reste à déterminer comment on peut obtenir les orbitales spatiales fi servant à construire le déterminant de Slater ; celles-ci étant des orbitales moléculaires (construites sur une base de fonctions qui reste à déterminer) dans le cas des systèmes polyatomiques. L'énergie moyenne du système s'obtient aisément après quelques manipulations mathématiques sur l'expression générale (3) en utilisant une fonction d'onde Y de la forme Slater. On obtient alors une expression pour l'énergie électronique moyenne (où l'on somme sur les n orbitales électroniques) :

où :

Dans l'expression ci-dessus, le terme Hii représente l'énergie d'un électron situé dans une orbitale moléculaire fi placé dans le champ des noyaux ; ce terme est multiplié par deux car il y a 2 électrons par orbitales (pour un système à « couches fermées »).
Les intégrales Jij et Kij sont respectivement appelées intégrales de Coulomb et intégrales d'échange ; l'intégrale de Coulomb a un équivalent en mécanique classique, alors que l'intégrale d'échange provient de la nécessité d'antisymétriser la fonction d'onde. Les intégrales de Coulomb et d'échange décrivent les interactions entre électrons. Jij représente l'interaction coulombienne moyenne entre deux électrons situés dans les orbitales fi et fj, sans tenir compte de leur spin. L'intégrale d'échange Kij réduit l'intéraction coulombienne entre deux électrons situés dans les orbitales fi et fj ayant des spins parallèles. Ce terme est une conséquence directe du principe de Pauli et conduit à une valeur d'énergie Ee plus basse, donc à une stabilisation. Par l'intermédiaire de l'intégrale d'échange on introduit ainsi une corrélation électronique entre électrons ayant des spins parallèles, c'est-à-dire que deux tels électrons ne peuvent pas se mouvoir indépendamment l'un de l'autre. On constate toutefois que ce modèle n'est pas apte à rendre compte de la corrélation entre électrons ayant des spins antiparallèles.
Nous nous proposons maintenant de résoudre l'équation de Schrödinger électronique (3) avec une fonction d'onde Y qui a la forme d'un déterminant de Slater afin de trouver l'expression des fonctions fi. Il est évident que ce déterminant ne peut pas être une solution exacte de l'équation de Schrödinger car une somme de termes monoélectroniques ne peut jamais être la solution d'une équation différentielle contenant des opérateurs biélectroniques. On doit, par conséquent, utiliser le principe variationnel [6].
En utilisant cette idée, Fock et Slater ont développé de façon simultanée et indépendante ce qui est maintenant connu sous le nom d'équations de Hartree-Fock [7]. Le principe variationnel dit qu'étant donnée une fonction d'onde d'essai  de la forme d'un déterminant de Slater, on peut montrer que l'on a toujours :
de la forme d'un déterminant de Slater, on peut montrer que l'on a toujours :

où E0 est l'énergie de la solution exacte  . La « meilleure » fonction d'onde de type déterminant de Slater sera donc obtenue en faisant varier tous les paramètres qu'elle contient, jusqu'à ce que l'on obtienne l'énergie la plus basse. Cela revient à minimiser la quantité
. La « meilleure » fonction d'onde de type déterminant de Slater sera donc obtenue en faisant varier tous les paramètres qu'elle contient, jusqu'à ce que l'on obtienne l'énergie la plus basse. Cela revient à minimiser la quantité  . En se rappelant qu'au cours de la minimisation, la fonction d'essai doit respecter la condition de normation
. En se rappelant qu'au cours de la minimisation, la fonction d'essai doit respecter la condition de normation  , le problème revient alors à faire une minimisation avec contrainte que l'on résout par la méthode des « multiplicateurs de Lagrange ».
, le problème revient alors à faire une minimisation avec contrainte que l'on résout par la méthode des « multiplicateurs de Lagrange ».
Soit une fonction G dépendante de plusieurs fonctions inconnues telle que :

où Sij provient de la condition d'orthonormalité : 
eij sont les multiplicateurs de Lagrange supposés réels
On a alors :

et on obtiendra les points stationnaires de la fonction G, au premier odre, en résolvant l'équation :
dG = 0
La variation infinitésimale dG est obtenue en faisant varier d'une quantité infinitésimale chaque orbitale fi, ce qui revient à remplacer fi par fi+dfi et fj par fj+dfj. On aura alors :

Après quelques manipulations mathématiques, il est possible de se ramener à un système d'équations différentielles, les « équations de Hartree-Fock » :

avec :

h(1) est l'opérateur qui prend en compte l'énergie cinétique de l'électron 1 et son énergie potentielle d'interaction avec le noyau A. Les termes J et K ont été définis précédemment. Il faut encore noter que l'opérateur K est non-local car, comme le montre l'expression ci-dessus, il dépend de la valeur de f(1) sur tout l'espace.
On constate ici que les opérateurs J et K s'expriment en fonction des solutions f de l'équation (8). On se trouve donc en présence d'un ensemble de N équations monoélectroniques non linéaires qu'il faudra résoudre par un processus itératif : à partir d'un jeu de spin-orbitales fi d'essai on calcule l'opérateur

pour en déduire ensuite un nouveau jeu de fonctions fi. Ce processus est nommé auto-cohérent.
Il est possible de montrer qu'il existe une transformation orthogonale des fi amenant la matrice des multiplicateurs de Lagrande eij à sa forme diagonale. En appliquant cette transformation à nos orbitales fi, on est apparemment conduit à un problème de valeurs propres puisque les équations (8) s'écrivent alors sous la forme :

Ici ei est l'énergie de l'orbitale i et F est l'opérateur monoélectronique de Fock donné par :

Ce système d'équations ne prend en compte que les orbitales spatiales fi. La seule référence au spin est faite lors du remplissage des orbitales où deux électrons seront placés par orbitale spatiale (principe de complémentarité appelé « aufbau »).
Les équations de Hartree-Fock sont un jeu d'équations intégro-différentielles couplées, et ne peuvent être résolues que par une méthode itérative. Le couplage se constate par le fait que les intégrales Jij et Kij sont définies en fonction des orbitales fi et fj, ce qui veut dire que pour déterminer F(1) dans (10) on a besoin de connaître les autres orbitales fj.
Pour résoudre ces équations, un jeu d'orbitales d'essai est donc choisi : l'opérateur de Fock est ensuite construit et le système d'équations (9) est résolu de façon à obtenir un nouveau jeu d'orbitales. Cette procédure est appelée « méthode à champ auto cohérent » (SCF = Self Consistent Field), car les itérations sont continuées jusqu'à ce que le champ électrostatique ressenti par un électron (champ provoqué par les autres électrons dans les autres orbitales) reste stationnaire.
Ces équations peuvent s'interpréter comme étant des équations de Schrödinger pour des électrons évoluant dans le champ des noyaux et des autres électrons du système, et dont les valeurs propres sont les énergies monoélectroniques ei associées aux fonctions propres, les spin-orbitales. Il reste maintenant à expliciter la forme des spin-orbitales fi pour résoudre les équations de Hartree-Fock.
2.1.5 L'approximation LCAO et les équations de Hartree-Fock-Roothaan
Nous avons vu que les orbitales moléculaires optimales s'obtiennent en résolvant un ensemble d'équations différentielles non linéaires (ne pouvant être résolues rigoureusement que pour des atomes dans l'hypothèse d'une distribution électronique globale sphérique). Cette technique conduit à une tabulation des orbitales, ce qui les rend inadéquates pour un bon nombre d'applications. Si l'on désire obtenir des spin-orbitales moléculaires sous une forme analytique, on doit se résigner à résoudre de manière approchée les équations de Hartree-Fock en choisissant pour orbitales moléculaires des combinaisons linéaires d'orbitales atomiques.
L'approximation LCAO proposée par Mulliken en 1941 [8] consiste à construire un jeu limité d'orbitales atomiques (OA) cm qui constituera une base sur laquelle seront développées les orbitales moléculaires fi (seule la partie spatiale des spin-orbitales est considérée ici). En essayant de résoudre les équations de Hartree-Fock pour des molécules, Hall, et indépendamment Roothaan, ont démontré qu'en introduisant un jeu de fonctions spatiales connues, les équations intégro-différentielles peuvent alors être transformées en un système d'équations algébriques et ainsi être résolues en utilisant la méthode habituelle des matrices [9]. Les nouvelles équations que l'on obtient dans cette approximation sont les équations de Hartree-Fock-Roothan.
Si l'on considère un ensemble de m orbitales atomiques (cl,cm,cn,cr, ...) servant de base au développement des m orbitales moléculaires fi(r) d'un système à couches fermées comportant 2n électrons, les orbitales moléculaires seront exprimées comme une combinaison linéaire de ces m fonctions spatiales mono-électroniques atomiques :

Les cmi sont les coefficients des orbitales moléculaires développées sur les fonctions de base. En toute rigueur le développement devrait être infini. Dans la pratique, il est clairement impossible de construire une base infinie d'orbitales. Par convention les OA sont centrées sur les atomes (d'où leur nom) et le symbole m correspond à l'atome sur lequel se trouve l'orbitale c. Il faut encore remarquer que malgré le terme « d'orbitales atomiques », celles-ci ne sont pas toujours les orbitales auto-cohérentes de l'atome isolé. Par cette méthode, les orbitales fi sont délocalisées sur l'ensemble de la molécule et pour cette raison elles s'appelleront « orbitales moléculaires ». La terminologie généralement admise pour désigner des orbitales moléculaires (OM) obtenues par l'optimisation des coefficients des fonctions de base atomiques qui sont des combinaison linéaires d'orbitales atomiques (LCAO) est LCAO-MO. Les orbitales moléculaires doivent, en outre, respecter les conditions de normation et d'othogonalité mutuelle que l'on écrit :

où dij est le symbole de Kronecker et Smn est communément appelée intégrale de recouvrement des orbitales cm et cn, et s'écrit :

Ce développement, appliqué aux équations de Hartree-Fock, conduit aux équations de Hartree-Fock-Roothan auxquelles on applique une fois encore le principe variationnel : on minimise l'énergie totale e par rapport aux coefficients du développement et l'on obtient alors les équations :

i = 1,2,...m étant les coefficients des orbitales moléculaires, et m = 1,2,...,m étant les coefficients des orbitales atomiques. On aura les termes suivants :

et  est la matrice de population pour ce système à couches fermées.
est la matrice de population pour ce système à couches fermées.
Le choix de la base constituée par les orbitales atomiques cm est fondamental, car il joue un rôle important, tant sur la précision des résultats, que sur les temps de calculs nécessaires pour les obtenir, comme il sera vu plus loin dans ce chapitre.
La résolution de ce système d'équations passe par l'annulation d'un déterminant construit sur les m équations à m+1 inconnues (les coefficients Cmi et les ei relatifs), ce qui conduit à l'équation séculaire du système étudié :
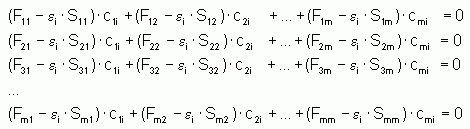
Sa résolution consiste alors à développer ce déterminant et à en trouver les racines (les ei) qui l'annulent. Chaque racine sera ensuite injectée à tour de rôle dans les équations de Hartree-Fock-Roothaan afin d'en obtenir les coefficients Cmi :

Le système n'est linéaire qu'en apparence car les éléments de matrice Fmn sont quadratiques dans les Cmi. Toutefois, pour pouvoir le résoudre on suppose qu'il est linéaire et on travaille de façon auto-cohérente. On remarque aussi que contrairement aux équations intégro-différentielles de Hartree-Fock, le système d'équations (12) est un système d'équations algébriques. Elles peuvent donc se ramener à l'équation séculaire, écrite dans sa forme générale déterminantale :

qui peut aussi s'écrire sous la forme matricielle suivante :
FC = SCE (13)
Les programmes de calculs travaillent généralement sous forme matricielle, ce qui évite de devoir résoudre des équations du nième degré (où n est le nombre de fonctions de base); ces équations, après transformation orthogonale, deviennent alors :

ce qui n'est rien d'autre qu'une équation aux valeurs propres et vecteurs propres, facilement résolvable par les ordinateurs. C est une matrice carrée des coefficients du développement et E est le vecteur des énergies.

L'équation ci-dessus est résolue d'une manière analogue à la résolution des équations de Hartree-Fock. Un premier essai est fait en utilisant des valeurs approchées pour les coefficients Cmi, la matrice de Fock est construite, puis elle est diagonalisée pour obtenir de nouveaux coefficients et de nouvelles énergies. Les nouveaux coefficients sont ensuite utilisés pour construire une nouvelle matrice de Fock et la procédure est répétée jusqu'à convergence des énergies ou des coefficients (dont le seuil est à fixer). L'énergie totale du système sera ensuite donnée par l'équation :

avec les éléments Pmn et Hmn précédemment définis.
Pour terminer, il faut encore remarquer que comme l'opérateur F est construit à partir de fonctions d'onde qui sont des approximations de celles de Hartree-Fock, il ne peut constituer qu'une forme approchée de l'hamiltonien de Hartree-Fock ; le système d'équations de Hartree-Fock-Roothaan ne constitue donc qu'une approximation des « vraies » équations de Hartree-Fock. La terminologie, « énergie Hartree-Fock » pour désigner le résultat de ces équations est donc abusive. En effet, si la base des OA était infinie, l'énergie E serait l'énergie de Hartree-Fock exacte, mais il n'en est rien. Les orbitales moléculaires obtenues dans l'approximation LCAO-MO ne sont donc que des approximations de celles de Hartree-Fock. Par convention, cependant, et sauf indication explicite, l'énergie issue du traitement Roothaan est appelée « énergie Hartree-Fock ».
2.1.6 Les fonctions de base utilisées dans l'approximation LCAO-MO
Comme il l'a été dit, le choix de la base de fonctions représentant les orbitales atomiques est important car il peut influencer tant la précision des résultats obtenus que les temps de calculs.
On distingue plusieurs types de bases d'orbitales atomiques : pour les bases minimales on choisit pour orbitales atomiques celles qui sont effectivement occupées à l'état fondamental du système en y ajoutant les orbitales inoccupées de la couche de valence. Chaque orbitale cm n'est décrite que par une seule fonction (la fonction 1s de l'hydrogène, par exemple).
Les bases étendues sont composées de la base minimale, où chaque orbitale est décrite par deux fonctions, à laquelle sont ajoutées un certain nombre d'orbitales situées au-delà de la couche de valence des différents atomes; celles-ci sont appelées orbitales de polarisation (pour l'hydrogène: 1s, 1s', 2px, 2py et 2pz).
Les bases de valence ne comprennent quant à elles que les orbitales de la couche de valence de chaque atome et en général une seule fonction de base par orbitale. Les électrons des couches internes (dits électrons de coeur) ne sont pas décrits explicitement dans ce type de base, mais un potentiel reproduit leur effet.
Il semblerait naturel d'utiliser, comme orbitales atomiques, des fonctions de Hartree-Fock obtenues en résolvant l'équation du champ auto-cohérent pour les atomes libres. Leur défaut essentiel est de ne pas posséder de forme analytique, de sorte que le calcul des intégrales moléculaires est considérablement alourdi. Pour ces raisons, on a préféré, historiquement, la base de fonctions de Slater, qui sont de bonnes approximations des orbitales de Hartree et qui s'écrivent dans leur forme générale [10] :

(n,l,m) sont les nombres quantiques principal, azimutal et magnétique, (r,q,f) sont les coordonnées sphériques définissant la position de l'électron, a est une constante déterminée à l'aide de règles empiriques, visant à reproduire au mieux le comportement des orbitales hydrogénoïdes, et Ui,m(q,f) sont les harmoniques sphériques des parties angulaires des solutions de l'équation de Schrödinger pour les atomes de type hydrogénoïdes.
Par rapport aux parties radiales hydrogénoïdes qui sont de meilleures solutions du problème atomique, les fonctions de Slater ont le désavantage de ne pas avoir de noeuds radiaux et, pour toutes celles de type s (sauf la 1s), d'être nulles à l'origine, ce qui n'est pas le cas des hydrogénoïdes. On résout pratiquement ces difficultés en utilisant le plus souvent des combinaisons linéaires de fonctions de Slater comme fonctions de bases atomiques, ce qui permet d'obtenir des représentations précises des orbitales atomiques de Hartree-Fock-Roothaan. Toutefois, si l'on construit une base LCAO de ce type pour un calcul moléculaire, le calcul des intégrales biélectroniques sera particulièrement difficile. La raison en est que le produit de deux orbitales de Slater situées sur des centres différents ne peut être exprimé simplement par une seule fonction. On préfère donc en général utiliser des fonctions de Gauss cartésiennes. Ces fonctions, proposées par Boys [11], sont des puissances de x,y,z multiplié par  , a étant une constante déterminant l'extension radiale de la fonction :
, a étant une constante déterminant l'extension radiale de la fonction :

où i,j,k sont des nombres entiers simulant les nombres quantiques n,l,m. N est le facteur de normalisation et x est l'exposant de la gaussienne.
Par exemple :

Les gaussiennes sont des fonctions très populaires en chimie quantique, spécialement pour les méthodes ab initio, car le produit de deux gaussiennes  centrées sur deux atomes A et B différents peut s'écrire à l'aide d'une seule gaussienne centrée en un point situé sur le segment AB. Le calcul des intégrales biélectroniques en ressort ainsi considérablement simplifié.
centrées sur deux atomes A et B différents peut s'écrire à l'aide d'une seule gaussienne centrée en un point situé sur le segment AB. Le calcul des intégrales biélectroniques en ressort ainsi considérablement simplifié.
Bien que les fonctions de Slater soient peu commodes d'utilisation pour les calculs numériques, elles présentent l'avantage de décrire raisonnablement les orbitales atomiques. Les bases gaussiennes ont, par contre, une assez mauvaise représentation des orbitales atomiques car elles n'ont pas un comportement exact à l'origine (dérivée devant être nulle), ni aux grandes distances (décroissance trop rapide avec r). Pour compenser la représentation incomplète des orbitales atomiques des fonctions gaussiennes, on utilise donc des combinaisons linéaires de gaussiennes comme fonctions de base. Ces fonctions sont appelées « fonctions gaussiennes contractées ». Il faut en général utiliser trois fonctions gaussiennes pour que l'ajustement des parties radiales soit satisfaisant. On aura par exemple :

où d1, d2, d3 sont les coefficients fixes de cette combinaison linéaire appelée « STO-3G ».
Il existe bon nombre de bases de gaussiennes possibles. Les plus communément utilisées sont celles qui ont été développées par Pople et collaborateurs [12]. La plus simple est la base STO-3G, aussi appelée « base minimale ». Le sigle « 3G » signifie que les orbitales de type Slater (STO) sont représentées par trois fonctions gaussiennes. Le niveau suivant développé par Pople comprend les bases split-valence telles que 3-21G, 4-31G et 6-31G, où le premier chiffre représente le nombre de gaussiennes utilisées pour représenter les orbitales de coeur. Les orbitales de valence y sont représentées par deux fonctions qui sont composées du nombre de gaussiennes indiqué dans la seconde partie de la dénomination de la base. Ainsi la base 6-31G du carbone, par exemple, utilisera six gaussiennes pour représenter l'orbitale 1s, trois gaussiennes pour l'orbitale 2s et 1 gaussienne pour représenter les orbitales 2p.
Pour une plus grande flexibilité on peut encore rajouter des fonctions de polarisation. La dénomination la plus ancienne est l'ajout d'un astérisque sur la base en question (par exemple 6-31G*), et dans une désignation plus récente, le caractère de la fonction ajoutée est explicitement donné : 6-31G(d). La base 6-31G* ou 6-31G(d) signifie ainsi qu'un jeu de fonctions d a été ajouté à tous les atomes (sauf H) dans la molécule, alors que 6-31G** ou 6-31G(p,d) signifie qu'un jeu de fonctions p a été ajouté aux hydrogènes et que des fonctions d ont été ajoutées aux autres atomes.
2.1.6.1 Les pseudopotentiels de coeur
Tous les électrons d'un atome ne jouent pas le même rôle. Ceux des couches internes (électrons de coeur) ne participent pas directement aux liaisons chimiques, alors que les électrons de la couche de valence sont les plus actifs. Il est donc parfois avantageux de remplacer les électrons de coeur par des potentiels effectifs ; la dimension du déterminant (6) en est ainsi réduite, en tenant compte de l'effet des orbitales de coeur par l'ajout de termes supplémentaires dans l'hamiltonien agissant sur cet espace réduit. En ne traitant explicitement que les électrons de valence on ne perd, en effet pratiquement aucune information sur les propriétés physico-chimiques des molécules, mais on réduit de façon significative le volume des calculs à effectuer, surtout si le système étudié contient des atomes lourds.
Dans le formalisme des pseudopotentiels de coeur les électrons des couches internes sont simulés par un opérateur monoélectronique appelé « pseudopotentiel » et l'un des avantages supplémentaires est que les effets relativistes peuvent être pris en compte dans le pseudopotentiel lui-même, et de ce fait un programme moléculaire non relativiste pourra être utilisé pour le calcul de molécules contenant des atomes des quatrième et cinquième lignes de la classification périodique.
2.1.7 Les méthodes de calculs utilisant l'approche Hartree-Fock
Les nombreuses méthodes de calculs fondées sur l'approximation de Hartree-Fock utilisent généralement l'approximation LCAO-MO comme point de départ. Les méthodes non-empiriques (ou ab initio) effectuent une résolution rigoureuse de ces équations en calculant toutes les intégrales à deux électrons. Les méthodes semi-empiriques négligent quant à elles un grand nombre de ces intégrales, et calculent les autres de manière approchée en faisant intervenir des paramètres ajustables déterminés empiriquement.
2.1.8 Le traitement de la corrélation électronique : les méthodes post Hartree-Fock
L'énergie Roothaan est égale à l'énergie Hartree-Fock dans le cas où la base de fonctions utilisée est infinie. Dans la théorie Hartree-Fock, l'énergie la plus basse pouvant être obtenue est EHF, c'est la limite Hartree-Fock. Or, cette théorie est approximative ; elle néglige, comme il l'a été dit, l'énergie de corrélation des électrons. Les électrons de spin opposés (particulièrement ceux situés dans des orbitales ayant des parties spatiales similaires) exercent, en effet, les uns sur les autres des forces répulsives dépendant de leurs positions instantanées. Or, dans le modèle des particules indépendantes de Hartree-Fock, cet effet est en partie négligé puisque l'on suppose que chaque électron se trouve dans le champ moyen crée par tous les autres. La contribution à l'énergie totale de cette interaction interélectronique d'origine quantique est faible, mais elle devient importante lorsque de petites différences d'énergie doivent être calculées.
D'après Löwdin [13], l'énergie de corrélation d'un système correspond à la différence entre l'énergie Hartree-Fock et l'énergie exacte non-relativiste du système :

Le modèle Hartree-Fock est certes très utile pour prédire certaines propriétés atomiques ou moléculaires, mais les méthodes post Hartree-Fock sont parfois nécessaires pour retrouver l'énergie de corrélation, car un traitement de la corrélation électronique plus poussé peut se révéler essentiel pour l'obtention de certaines propriétés atomiques ou moléculaires. La recherche des fonctions d'onde sera donc rendue plus compliquée, et pour ce faire, plusieurs méthodes ont été proposées, comme il l'a déjà été mentionné en introduction au chapitre I. Deux grandes catégories de méthodes existent actuellement : les méthodes à référence unique et les méthodes multiréférencées.
La fonction d'onde HF ne décrit pas correctement le comportement des électrons à proximité du noyau et surestime la probabilité de trouver deux électrons proches l'un de l'autre. Ces effets de corrélation à courte distance sont dus au trou de Coulomb [14] et l'énergie de corrélation qui en découle est appelée, comme déjà dit, « corrélation dynamique ». Les effets de corrélation à longue distance contribuent, quant à eux, à l'énergie de « corrélation non dynamique » (ou statique) et à cause de ces effets, les calculs HF ont tendance à sous-estimer les longueurs de liaison. Lorsque ces effets sont faibles, la fonction d'onde HF fournit une bonne description du système et pour récupérer l'énergie de corrélation, des méthodes post-HF basées sur une référence unique suffisent.
Par chance, cette situation est la plus répandue (systèmes à l'état fondamental proche de l'équilibre) ; par contre, dans les autres situations, la description mono-déterminantale de la théorie HF est insuffisante (états excités, molécules proches de la dissociation ou états électroniquement quasi-dégénérés). Dés lors il faut utiliser des méthodes post-HF multiréférencées (MCSCF, MRCI) qui ne seront pas décrites ici.
2.1.8.1 Les méthodes à référence unique : l'approche perturbative Møller-Plesset
La méthode de perturbation Møller-Plesset [23] est une adaptation aux systèmes polyélectroniques de la théorie, plus générale, développée par Rayleigh et Schrödinger et connue sous le nom de théorie des perturbations à plusieurs corps (MBPT).
Dans la théorie de perturbation de Møller-Plesset, l'hamiltonien total est séparé en deux : une partie qui a les fonctions propres et les valeurs propres du déterminant Hartree-Fock, et une partie perturbée V. L'énergie est alors exprimée comme une somme de ces deux contributions :

F étant l'opérateur de Fock, et V étant le potentiel de corrélation défini par :

On connaît déjà les solutions de l'équation :

La théorie des perturbations stipule que si V est petit par rapport à F, on peut alors développer l'opérateur H = F+lV en série de Taylor selon l, d'où :

Et on peut ainsi montrer que :

La perturbation la plus couramment utilisée est la perturbation au deuxième ordre. Elle est connue sous le nom de « MP2 ». Cette méthode permet de récupérer une grande partie de l'énergie de corrélation. Elle n'est cependant pas variationnelle et est basée sur une référence unique (la fonction d'onde de Hartree-Fock). Cette méthode est très efficace et requière dans la pratique des temps de calculs acceptables, proportionnels à N5, où N est le nombre d'électrons du système étudié.
Il existe d'autres méthodes à référence unique telle que l'interaction de configuration (CI) ou la méthode « coupled cluster » (CC). Il faut encore remarquer que les autres types de méthodes post-HF, telles que l'interaction de configuration, sont des méthodes variationelles, contrairement à la méthode de Møller-Plesset, ce qui implique que si les énergies obtenues par CI ne peuvent pas être inférieures à l'énergie réelle du système, les énergies calculées par la méthode de Møller-Plesset peuvent l'être.
Les techniques post-HF sont en général très efficaces pour retrouver l'énergie de corrélation, mais cependant à l'heure actuelle elles sont, pour la majeure partie d'entre-elles, trop lourdes pour être applicables à des systèmes dont le nombre d'atomes est grand. Il s'est ainsi parallèlement développé à ces techniques un modèle alternatif qui a atteint le statut de théorie à la fin des années 60. La théorie de la fonctionnelle de la densité (DFT) est actuellement la seule permettant l'étude de systèmes chimiques de grande taille avec la prise en compte des effets de la corrélation électronique de manière satisfaisante.
2.2 La théorie de la fonctionnelle de densité (DFT)
2.2.1 Fondements de la théorie
La théorie de la fonctionnelle de la densité est basée sur le postulat proposé par Thomas et Fermi qui dit que les propriétés électroniques peuvent être décrites en terme de fonctionnelles de la densité électronique, en appliquant localement des relations appropriées à un système électronique homogène [15]. Thomas et Fermi ont utilisé leur théorie pour la description d'atomes, mais le manque de précision, ainsi que l'impossibilité de traiter des systèmes moléculaires en ont fait un modèle trop simpliste lorsqu'il a été proposé.
Hohenberg et Kohn, en 1964 [16], ont repris la théorie de Thomas-Fermi et ont montré qu'il existe une fonctionnelle de l'énergie E[r(r)] associée à un principe variationnel, ce qui a permis de jeter les bases de la théorie de la fonctionnelle de la densité. Des applications pratiques ont ensuite été possibles grâce aux travaux de Kohn et Sham (KS) [17] qui ont proposé, en 1965, un set d'équations monoélectroniques analogues aux équations de Hartree-Fock à partir desquelles il est en principe possible d'obtenir la densité électronique d'un système et donc son énergie totale.
Fonctionnelle et dérivée fonctionnelle sont des entités mathématiques de première importance dans la théorie DFT. Mathématiquement, on désigne par « fonctionnelle » une entité qui fait correspondre un nombre à chaque fonction provenant d'une classe définie. En d'autres termes, c'est une fonction de fonction. La notation d'une fonctionnelle est F[f(r)], où r est une variable de la fonction f. La dérivée fonctionnelle est la quantité  telle que :
telle que :

L'équation (15) représente la série coupée jusqu'au terme linéaire de dr. Comme il va l'être vu plus avant, il existe une correspondance biunivoque entre la densité électronique d'un système et le potentiel externe v(r). La densité électronique r(r) constitue la grandeur fondamentale de la DFT, et les termes de l'hamiltonien électronique (4) utilisé en début de ce chapitre peuvent s'écrire en fonction de matrices densité, grandeurs qui généralisent la notion de densité électronique.
La densité électronique r(r1) de l'électron 1, de coordonnées r1, est en fait l'élément diagonal d'une matrice densité 1 r1(r1',r1). Si y est la spin-orbitale donnant la densité r(r1), on peut alors calculer r(r1) d'après l'expression :

où les si sont les coordonnées de spin et les ri sont les coordonnées d'espace. r1 est donc une « matrice densité d'ordre 1 » [25].
De la même manière, on défini une « matrice densité d'ordre 2 » r2(r1'r2', r1r2) dont l'élément de matrice diagonal est r2(r1r2,r1r2) = r2(r1r2) et dont l'expression est :

Il est important de noter que l'intégrale sur tout l'espace de r(r1) donne le nombre d'électrons N total du système, tandis que la matrice densité d'ordre 2 intègre sur le nombre de paires d'électrons 
Plus généralement, on peut construire une matrice que nous appellerons matrice densité d'ordre p, et telle que :

où  correspond au coefficient binômial.
correspond au coefficient binômial.
Dans le cadre de la description des propriétés régies par des intéractions interélectroniques, les matrices densité d'ordre 1 et 2 suffisent.
Avec ces nouvelles grandeurs il est maintenant possible de réécrire chacun des composants d'énergie provenant de l'hamiltonien (4) :

On constate que le terme Vee[r] est composé de deux parties; la première correspond à l'interaction coulombienne classique J[r], et la seconde partie dite non-classique est appelée « énergie d'échange et de corrélation ».
2.2.2 Les théorèmes de Hohenberg et Kohn
Les deux théorèmes de Hohenberg et Kohn formulés en 1964 [16] ont permis de donner une cohérence aux modèles développés sur la base de la théorie proposée par Thomas et Fermi à la fin des années 30.
2.2.2.1 Premier théorème
Le premier théorème démontre que pour un système électronique décrit par un hamiltonien H de la forme de celui utilisé en début de ce chapitre (4), le potentiel externe v(r) est déterminé, à une constante additive près, par la densité électronique r(r) du système. Comme r(r) détermine le nombre d'électrons, la densité nous permet donc d'accéder à toutes les propriétés électroniques relatives à l'état fondamental du système.
On peut alors utiliser la densité électronique comme variable de base pour la résolution de l'équation de Schrödinger électronique. Etant donné que r(r) est liée au nombre d'électrons du système, elle peut en effet également déterminer les fonctions propres Y de l'état fondamental ainsi que toutes les autres propriétés électroniques du système ; si N est le nombre d'électrons du système, on a que :

Connaissant la densité électronique r(r) d'un système, on a donc accès au nombre d'électrons, au potentiel externe, ainsi qu'à l'énergie totale Ev[r]. Celle-ci peut s'écrire sous la forme :

où FHK[r] = T[r]+Vee[r] est la fonctionnelle universelle de Hohenberg et Kohn.
FHK[r] est une fonctionnelle prenant en compte tous les effets interélectroniques ; elle est indépendante du potentiel externe, et elle est donc valable quelque soit le système étudié. La connaissance de FHK[r] permet l'étude de tous les systèmes moléculaires, malheureusement la forme exacte de cette fonctionnelle est à l'heure actuelle loin d'être connue, et il faut avoir recours à des approximations.
2.2.2.2 Deuxième théorème
Le second théorème établit le principe variationnel de l'énergie Ev[r]. Pour une densité électronique d'essai,  , telle que
, telle que  et
et  , on a toujours
, on a toujours 
La condition pour qu'une fonctionnelle telle que Ev[r] admette un extremum est que sa dérivée fonctionnelle s'annule. D'après la définition :

La relation dEv = 0 est donc vérifiée si :

La résolution du problème consiste dés lors à chercher à minimiser Ev[r] avec la contrainte  . On résout le problème une fois encore par l'utilisation de multiplicateurs de Lagrange. Soit :
. On résout le problème une fois encore par l'utilisation de multiplicateurs de Lagrange. Soit :

La contrainte devient G[r] = 0, et si on introduit une fonctionnelle auxiliaire A[r] telle que :

où m est un multiplicateur de Lagrange, le problème se résume alors à résoudre :

soit :

Il faut alors calculer la dérivée fonctionnelle de A[r] :

Si l'on remplace l'expression ci-dessus dans l'expression de dA[r], il vient :

et il reste à calculer la dérivée fonctionnelle de Ev[r]. D'après les équations (15) et (19), il vient :

En remplaçant cette dernière équation dans l'expression (20), on obtient l'équation fondamentale de la DFT, qui est une équation de type Euler-Lagrange :

où la quantité m est appelée « potentiel chimique » du système.
Les théorèmes de Hohenberg et Kohn ne donnent cependant aucune information sur la manière de trouver la fonctionnelle FHK[r], et il va donc falloir trouver une méthode adéquate pour traiter ce problème.
2.2.2.3 Formulation de la recherche par contrainte
Calculer la densité électronique de l'état fondamental en connaissant sa fonction d'onde est un problème trivial. Par contre, plusieurs fonctions d'onde différentes peuvent conduire à la même densité. Dés lors, connaissant la densité électronique de l'état fondamental, comment trouver la fonction d'onde correspondante ?
La réponse est donnée par la recherche par contrainte établie par Levy [18] qui généralise le deuxième théorème de Hohenberg et Kohn. Le principe variationnel établit que :

Cette minimisation peut être réalisée en deux temps :

Ainsi, on cherche les fonctions d'onde conduisant à cette densité et minimisant l'énergie parmi toutes les densités électroniques. On montre alors que le problème peut s'exprimer en fonction de la fonctionnelle universelle de Hohenberg et Kohn :

avec :

où Vee est l'énergie d'interaction interélectronique. La relation ci-dessus propose une recherche par contrainte de la densité électronique : la recherche de la fonction d'onde de l'état fondamental se fait uniquement parmi les fonctions d'onde conduisant à la densité r. Par conséquent, la fonctionnelle F minimise la valeur moyenne des opérateurs d'énergie T+Vee pour toutes les fonctions d'essai Y décrivant la densité r.
2.2.3 La méthodologie de Kohn-Sham
La fonctionnelle de Hohenberg et Kohn contient une composante d'énergie cinétique T[r] et une composante d'énergie potentielle Vee[r]. Cette dernière peut, comme il l'a déjà été dit, elle-même se scinder en une partie classique (la répulsion coulombienne), notée J[r], et une partie d'origine quantique, K[r]. Thomas et Fermi avaient proposé une approximation de T[r], mais celle-ci, comme il l'a été dit, s'est révélée être insuffisante pour décrire de manière satisfaisante l'énergie cinétique des systèmes électroniques. Kohn et Sham ont proposé en 1965 [17] de calculer une énergie cinétique approchéeTs[r] en introduisant les orbitales.
Cette méthode, plus indirecte, est donc basée sur l'utilisation d'orbitales qui permettent d'évaluer avec une bonne précision l'énergie cinétique ; une faible correction étant apportée dans un second temps. La formulation exacte de l'énergie cinétique pour l'état fondamental est la suivante :

où les yi sont les spin-orbitales naturelles du système et ni est leur nombre d'occupation respectif. Le principe de Pauli impose la condition 0 Ł ni Ł 1 et selon la théorie de Hohenberg-Kohn, l'énergie cinétique T est une fonctionnelle de la densité électronique totale donnée par :

Pour un système où les électrons sont sujets à des interactions, il y a néanmoins un nombre infini de termes dans les expressions de T et de r.
Ces équations correspondent en fait au cas où ni = 1 pour N orbitales, et ni = 0 pour le reste. Cette condition n'est valable que pour les fonctions d'onde déterminantales décrivant un système à N électrons sans interactions. Afin d'avoir une unique décomposition en termes d'orbitales conduisant à une seule valeur exacte pour TS[r], Kohn et Sham ont proposé, par analogie avec la définition de la fonctionnelle universelle de Hohenberg et Kohn, un système de référence sans interactions, et l'énergie cinétique est calculée selon l'expression :
 pour les N orbitales
pour les N orbitalesla quantité T[r]-Ts[r] étant cependant faible.
A priori Ts[r] n'est pas l'énergie cinétique du système étudié ; Kohn et Sham ont reformulé le problème de manière à ce que le système de référence d'électrons non-intéragissant ait la même densité électronique que l'état fondamental du système étudié. Pour cela, ils ont réécrit la fonctionnelle F[r] de la manière suivante :
F[r] = Ts[r] + J[r] + Exc[r]
avec :
Exc[r] = T[r] - Ts[r] + Vee[r] - J[r]
La quantité Exc[r] est appelée « énergie d'échange-corrélation ». L'équation (21) devient alors :

avec le potentiel effectif Veff :

où vxc est le potentiel d'échange-corrélation, dérivée fonctionnelle de Exc[r] par rapport à r(r). L'équation (22) est exactement la même que celle de la théorie de Hohenberg et Kohn pour un système d'électrons non-intéragissant se déplaçant dans un potentiel effectif de la forme de veff(r).
En appliquant le principe variationnel, on obtient alors un ensemble d'équations du type Hartree-Fock que l'on résout par un processus intératif :

La densité électronique est ensuite obtenue par la sommation :

Pratiquement, on choisit une densité d'essai à partir de laquelle on calcule un potentiel effectif veff(r). En injectant veff(r) dans l'expression (23) on obtient une nouvelle densité électronique (24). La convergence est alors atteinte lorsque le potentiel effectif ne varie plus.
Ces équations sont analogues à celles obtenues par la méthode de Hartree-Fock, mais contiennent un potentiel local plus général Veff(r). Les théories quantiques vues dans ce chapitre (Hartree, Hartree-Fock et Kohn-Sham) conduisent toutes à un système d'équations mono-électroniques, mais le formalisme de Kohn-Sham permet néanmoins de tenir compte, de manière intrinsèque, de l'effet dû à l'échange et à la corrélation électronique.
Il faut encore ajouter que le terme Veff(r) ne contient pas d'opérateur de spin, et chaque solution pour ei est doublement dégénérée ; on a donc les deux cas suivants :

Pour le cas « closed-shell », on aura :

Pour un système à couches ouvertes, on aura par contre :

Cette condition de restriction découle directement de la théorie, alors que dans le cas Hartree-Fock elle était la conséquence de l'approximation orbitale de Hartree. Il faut noter que les orbitales utilisées dans l'équation de Kohn-Sham sont celles conduisant à un minimum pour l'énergie totale et sont obtenues de manière auto-cohérente. La signification physique de ces orbitales n'est cependant pas claire ; l'orbitale HOMO permet néanmoins d'obtenir la valeur du potentiel d'ionisation, sur la base du théorème de Janak.
Kohn et Sham ont donc permis à la DFT de devenir un outil efficace pour l'étude des systèmes chimiques. Actuellement, la très grande majorité des calculs DFT sont réalisés dans le cadre de ce formalisme ; les approximations qui vont brièvement être décrites ci-après s'inscrivent dans le cadre du formalisme de Kohn-Sham.
2.2.4 L'approximation locale LDA
La difficulté principale dans le développement du formalisme de Kohn-Sham réside dans la construction des fonctionnelles d'échange-corrélation. L'approximation locale dite « LDA » stipule qu'en première approximation la densité peut être considérée comme étant localement constante. On peut dés lors définir l'énergie d'échange-corrélation de la manière suivante :

où exc(r) est la densité d'énergie d'échange-corrélation.
Cette approximation découle directement du modèle du gaz homogène d'électrons. Par ailleurs, si l'on partitionne l'énergie d'échange-corrélation en deux (énergie d'échange ex et énergie de corrélation ec) telle que :
exc = ex + ec
on peut utiliser l'énergie d'échange proposée par Dirac [26] comme approximation de ex :

La fonctionnelle de corrélation la plus utilisée a été développée par Vosko, Wilk et Nusair en 1980 [27]. Ces auteurs ont utilisé les résultats de calculs Monte Carlo effectués par Ceperley et Alder pour ajuster une expression analytique de l'énergie de corrélation. Cette fonctionnelle est connue sous l'abréviation « VWN ».
Dans la pratique, la méthode LDA se montre plus performante que les calculs Hartree-Fock. On constate cependant qu'en général cette approximation a tendance à raccourcir les longueurs de liaison dans les molécules et, par conséquent, à surestimer les énergies de liaison. De plus, il est très fréquent que les barrières d'activation des réactions chimiques soient largement sous-estimées. Les fréquences de vibration sont par contre généralement en bon accord avec l'expérience (l'écart étant souvent inférieur à 5 %).
Depuis 1985 d'énormes efforts ont contribué à l'amélioration des fonctionnelles d'échange-corrélation. Ces travaux ont débouché sur une deuxième génération de fonctionnelles incluant l'inhomogénéité de la densité électronique : ces fonctionnelles prennent donc en compte la densité électronique ainsi que son gradient.
2.2.5 L'approximation des gradients généralisés GGA
La densité électronique d'un système est non seulement pas uniforme, mais peut même varier très rapidement dans l'espace (lorsqu'on passe d'une couche électronique à l'autre dans un atome, ou lorsqu'on passe d'un atome à l'autre dans une molécule). La première amélioration que l'on puisse apporter à la méthode LDA consiste donc à exprimer la fonctionnelle d'énergie d'échange-corrélation en fonction de la densité électronique et de son gradient.
Cette technique est appelée « approximation de l'expansion du gradient » (GEA). Elle se révèle efficace pour les systèmes dont la densité électronique ne varie que lentement. Pour les systèmes chimiques, il s'avère qu'elle donne des résultats moins bons que LDA. La solution consiste alors à réécrire l'expression d'échange-corrélation sous une forme similaire à LDA :

où excGGA est la densité d'énergie d'échange-corrélation. La difficulté réside dés lors dans la recherche d'expressions analytiques de excGGA.
De nombreuses fonctionnelles ont été developpées depuis, tant pour l'échange que pour la corrélation. Parmi les plus connues et les plus utilisées on peut citer les fonctionnelles d'échange de Becke (B88) [28] et de Perdew et Wang (PW91) [29]. Pour la corrélation, on dispose, entre autres, des fonctionnelles de Perdew (P86) [30], de Lee, Yang et Parr (LYP) [31] et de Perdew et Wang (PW91) [29]. Toutes ces fonctionnelles permettent une amélioration de l'estimation des énergies de liaison dans les molécules, ainsi que des barrières d'énergie par rapport à l'approximation locale LDA.
Il faut encore citer les fonctionnelles dites « hybrides », basées sur le formalisme de la connection adiabatique [32]. Le principe émerge de la question demandant s'il est possible d'utiliser l'échange de Hartree-Fock dans le formalisme de Kohn-Sham. La formule de la connection adiabatique justifie théoriquement la détermination de l'énergie d'échange HF à partir de l'énergie des orbitales Kohn-Sham. L'utilisation de la partie d'échange HF associée aux fonctionnelles GGA fournit des résultats comparables à ceux de l'approximation des gradients généralisés. La première fonctionnelle de ce type a été proposée par Becke, et contient 50 % d'échange HF ; c'est la fonctionnelle « half and half » [33]. Elle présentait l'inconvénient de contenir une trop forte proportion d'échange HF, et la fonctionnelle de ce type actuellement la plus utilisée est celle connue sous l'acronyme B3LYP [34]. Celle-ci est une fonctionnelle à trois paramètres combinant les fonctionnelles d'échange local, d'échange de Becke et d'échange HF, avec les fonctionnelles de corrélation locale (VWN) et corrigée du gradient de Lee, Yang et Parr.
Enfin, de nouveaux travaux ont récemment été entrepris afin de développer de nouvelles fonctionnelles ab initio sans paramètres. A l'heure actuelle, il n'existe qu'une seule fonctionnelle de ce type, élaborée par Perdew, Burke et Ernzerhof (PBE) [35], qui s'est montrée très efficace pour les calculs de géométries, de fréquences et d'énergies d'excitations électroniques.
2.2.6 Conclusion
Les développements théoriques ont permis de faire de la physique quantique appliquée à la chimie un outil indispensable associé à la chimie expérimentale.
Nous avons eu ainsi moyen, dans ce chapitre, de constater qu'au cours des années de développement de la chimie quantique deux voies se sont dégagées ; l'une aborde les problèmes en décrivant les systèmes par une fonction d'onde, l'autre le fait par le biais de sa densité électronique.
Les méthodes DFT souffrent cependant d'un manque de procédures systématiques qui permettent d'améliorer les fonctionnelles et les propriétés moléculaires calculées, ce qui n'est pas le cas avec les calculs ab initio pour lesquels il est à priori possible d'augmenter la qualité des résultats en augmentant le niveau de calculs ou la qualité de la base de fonctions. La seule limitation dans le cas ab initio étant naturellement liée au temps requis pour effectuer de tels calculs.
Il a été vu que ces deux méthodes peuvent parfois être utilisées conjointement, et il ne serait pas si surprenant que dans un futur proche, ces deux théories donnent naissance à une nouvelle théorie mixte dans laquelle la fonctionnelle d'énergie serait orbitalairement dépendante et non plus densité-dépendante [36].
2.3 Les méthodes de solvatation
2.3.1 La méthode du « continuum solvation »
La possibilité d'intégrer les effets dus au solvant pour le calcul des différentes propriétés des systèmes chimiques reste un challenge dans la chimie quantique, car cela implique l'intervention de la mécanique statistique et donc, l'ajout de difficultés d'ordre supérieur. La majorité des réactions chimiques et biologiques ont cependant lieu en solution, et le désir du chimiste théorique est donc celui de pouvoir posséder et utiliser des modèles permettant de tenir compte des effets dus au solvant.
Tomasi et Persico [42,45-46] ont proposé de diviser les différentes approches possibles du traitement des effets de solvant en quatre catégories :
- Equation d'état virielle, fonctions de corrélation
- Simulation de type Monte Carlo ou de dynamique moléculaire
- Traitements de type continuum
- Traitements moléculaires
La partie de théorie présentée ci-après ne se veut être qu'une introduction des différentes méthodes de solvatation ; pour plus de détails, l'excellent article de Tomasi et Persico [46] les passe en revue, tout en présentant les éléments mathématiques absents ici.
L'idée de modéliser les interactions électrostatiques dues au solvant en plaçant le soluté dans une cavité de taille définie date des travaux de Kirkwood [43] et Onsager sur les effets de solvatation sur les molécules polaires [44]. A partir de l'équation de Laplace (ou de Poisson), et sous certaines conditions limites, plusieurs modèles ont été par la suite proposés [41-50]. Dans cette approche, le soluté, traité de manière quantique, est placé dans une cavité entourée de molécules de solvant considérées comme un continuum. Ce modèle de continuum simple est le « modèle de la cavité d'Onsager », souvent dénommé « modèle SCRF », pour « Self-Consistent Reaction Field ».
Les modèles de type « continuum » impliquent toutes sortes de formes de cavité contenant le soluté, et le solvant se trouvant en-dehors est traité comme un milieu continu, caractérisé par quelques-unes seulement de ses propriétés comme sa constante diélectrique, par exemple. Le champ électrique produit par les particules chargées comprenant le soluté interagit alors avec ce milieu, produisant une polarisation, ce qui se reflète sur les fonctions d'onde du soluté.
La cavité représente la portion de l'espace où la densité de solvant est nulle et où le soluté prend place. Il est bien connu que la réponse d'un milieu diélectrique continu à toute distribution de charge consiste en une distribution de charge de surface à l'interface entre les deux. Le problème, non trivial, consiste dès lors à calculer cette charge de surface ; la partie la plus difficile étant de résoudre l'équation de Poisson avec une telle condition de manière analytique simple.

n(r) est le vecteur normal de la surface en un point r et E- (r) est le champ électrique total à l'intérieur de la surface en ce point. Les charges d'écran s devant se calculer de manière itérative (elles viennent s'ajouter aux cycles SCF de la molécule isolée), les méthodes incluant les effets de solvatation requièrent donc des temps de calculs plus longs.
L'approche la plus simple utilise des séries de Taylor pour représenter l'expansion classique multipolaire de la structure électronique. Cette expansion est tronquée au terme dipolaire et seules les interactions des charges atomiques et des dipôles avec le milieu sont donc prises en compte. L'approche SCRF utilise une cavité idéale (sphérique ou ellipsoïdale) qui permet une solution analytique pour le calcul de l'énergie d'interaction entre le multipôle du soluté et celui du continuum. Ces simplifications conduisent à des problèmes de précision, et cette méthode est très sensible au choix du rayon de la cavité, mais pas tant à la constante diélectrique (pour les solvants particulièrement polaires).
Ce modèle est implémenté en standard dans les programmes comme « GAMESS » ou le « GAUSSIAN » de la manière la plus simple possible :
- utilisation d'une cavité sphérique
- le potentiel électrostatique du soluté est représenté par sa charge (dans le cas d'un ion) ou par le moment dipolaire
L'interaction électrostatique du soluté ne peut être résumée en ces deux termes uniquement. Au final, la restriction de la cavité sphérique n'est donc pas représentative de la véritable forme du solvant, et seul dans les cas spéciaux de molécules particulièrement sphériques, le modèle SCRF peut se montrer suffisant.
Récemment, une approche de type continuum utilisant une généralisation du formalisme de Born pour l'interaction entre les charges atomiques partielles et le milieu diélectrique a été proposée [52]. Dans le principe, les monopoles centrés sur les atomes génèrent tous les multipôles requis pour représenter la distribution électronique.
Une méthode plus sophistiquée encore, dénommée « Polarizable Continuum Model » (PCM) a été développée par Tomasi et ses collaborateurs [45-47]; celle-ci permet de travailler avec des cavités de forme plus réaliste, avec une surface découpée en une sorte de mosaïque constituée de petits polygones sphériques. L'interaction électrostatique entre le soluté et le solvant est dans ce cas décrit par un ensemble de charges ponctuelles polarisables, placées au centre de chaque petit 'morceau' (tessera). Ce modèle est donc beaucoup plus versatile en termes de description réaliste de la cavité et plus précis en ce qui concerne l'énergie due à l'interaction électrique entre le soluté et le milieu environnant.
La méthode PCM place ainsi le soluté dans une cavité formée par l'union de sphères centrées sur chaque atome et le potentiel électrostatique du soluté est décrit par la production d'une charge apparente (de surface) sur la surface de la cavité, ce qui implique un plus grand réalisme pour l'interaction électrostatique. Le traitement par ordinateur divise la surface en de petits morceaux sur lesquels la charge (et sa contribution au gradient) est évaluée. Sur la base de différentes études, on a défini la taille de ces sphères comme ayant un volume équivalent à environ 1,2 fois le rayon de Van der Waals [45-50].
Le modèle COSMO-PCM (CPCM) représente quant à lui une approche différente basée sur l'implémentation du « Conductor like Screening Model (COSMO) of solvation » [49-50]. Dans le modèle COSMO, le milieu environnant est décrit par un conducteur et non plus par un milieu diélectrique (e = Ą), permettant de fixer les conditions limites initiales. Les termes d'énergie calculés en premier lieu pour le conducteur, sont ensuite divisés par un facteur de scaling décrit par la fonction :

où x est un facteur de correction empirique (fixé par comparaison avec les valeurs obtenues pour des cas analytiques simples impliquant un milieu diélectrique) et e est la constante diélectrique (ici de l'eau), ce qui permet de revenir au milieu diélectrique originel.
Cette technique simplifie les calculs d'interactions électrostatiques et les corrections sont effectuées à posteriori pour le comportement diélectrique. Les implémentations actuelles de ce modèle incluent le calcul de multipoles allant jusqu'aux hexadecapoles pour représenter la densité de charge de la molécule de soluté. Cette distribution induit à son tour une distribution de charge à la surface de la cavité et cela est pris en compte dans les cycles de calculs SCF, ce qui permet un traitement auto-cohérent pour les fonctions d'ondes moléculaires et les charges de la surface.
Il est généralement reconnu que les erreurs de cavité sont plus faibles dans la méthode COSMO-PCM (CPCM) que dans la méthode PCM seule, ceci étant dû à l'utilisation, dans COSMO, de conditions limites exprimées en termes de potentiel électrostatique plutôt qu'en termes de champ électrique.
Ces modèles ont cependant de nombreuses limitations ; l'une des plus importantes étant qu'ils ne permettent pas de tenir compte de l'aspect dynamique des effets entre le soluté et le solvant (liaisons hydrogène, par exemple). Malgré cela, ces méthodes de solvatation peuvent être utilisées (approche du continuum ; méthodes SCRF et PCM) afin d'améliorer les énergies et les géométries des espèces chimiques intervenant dans mécanismes réactionnels, par exemple.




















 H10. Les sept hydrogènes Hs permettent de saturer les liaisons O-Al et les hydrogènes Hn ont été ajoutés afin de rendre le système neutre.
H10. Les sept hydrogènes Hs permettent de saturer les liaisons O-Al et les hydrogènes Hn ont été ajoutés afin de rendre le système neutre.





























 est la distance M
est la distance M  O (active) à l'état de transition
O (active) à l'état de transition
 sont les longueurs de liaisons « normales » à l'état de transition
sont les longueurs de liaisons « normales » à l'état de transition


















