

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|



|
||||||||
The fluorescence decays of several exciplexes with partial charge transfer have been investigated in solvents of various polarity. The measured lifetimes are found to be in reasonable agreement with the activation enthalpy and entropy of exciplex decay obtained earlier from the temperature dependence of the exciplex emission quantum yields. For exciplexes with 9-cyanophenanthrene substantial contribution of the higher local excited state into the exciplex electronic structure is found and borrowed intensity effect enhances the exciplex emission rate constants. | ||||||||
|
||||||||
Free ion formation in acetonitrile is examined through transient photoconductivity for a set of ketones excited at different wavelengths. According to the photophysical parameters of the ketones and the incident photon energy, two mechanisms can be operative: triplet–triplet annihilation (bimolecular process) and/or photoionization (monomolecular biphotonic process). By using a tunable laser, excited state mediated photoionization was studied. From the threshold energy (Ethr) required for this process to occur, ionization potentials in solution (IS) were deduced and compared to the corresponding values in gas phase (IG). A simple energetic model enables the determination of the oxidation potential (Eox) of the ketones that are compared to the corresponding values obtained through electrochemical measurements. | ||||||||
|
 |
|||||||
Electronic energy transfer (EET) rate constants between a naphthalene donor and anthracene acceptor in [ZnL4a](ClO4)2 and [ZnL4b](ClO4)2 were determined by time-resolved fluorescence where L4a and L4b are the trans and cis isomers of 6-((anthracen-9-yl-methyl)amino)-6,13-dimethyl-13-((naphthalen-1-yl-methyl)amino)-1,4,8,11-tetraazacyclotetradecane, respectively. These isomers differ in the relative disposition of the appended chromophores with respect to the macrocyclic plane. The trans isomer has an energy transfer rate constant (kEET) of 8.7 × 108 s-1, whereas that of the cis isomer is significantly faster (2.3 × 109 s-1). Molecular modeling was used to determine the likely distribution of conformations in CH3CN solution for these complexes in an attempt to identify any distance or orientation dependency that may account for the differing rate constants observed. The calculated conformational distributions together with analysis by 1H NMR for the [ZnL4a]2+ trans complex in the common trans-III N-based isomer gave a calculated Förster rate constant close to that observed experimentally. For the [ZnL4b]2+ cis complex, the experimentally determined rate constant may be attributed to a combination of trans-III and trans-I N-based isomeric forms of the complex in solution. | ||||||||
|
||||||||
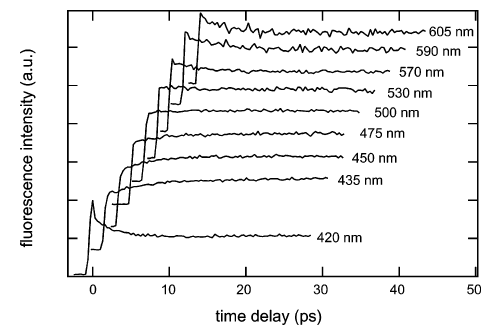
The charge recombination dynamics of geminate ion pairs formed by electron transfer quenching of cyanoanthracene derivatives by bromo- and iodo-anisole in acetonitrile was investigated using various ultrafast spectroscopic techniques. Without a heavy atom, the only charge recombination pathway is that leading to the neutral ground state. With heavy atom substituted anisoles, charge recombination to the local triplet state of the excited precursor is observed. Time constants for triplet charge recombination ranging from 400 ps to less than 10 ps, depending on the heavy atom and on the energy gap between the ion pair and the triplet state, have been measured. This heavy atom effect was observed with ion pairs formed upon electron-transfer quenching with driving force going from −0.15 to −0.6 eV, suggesting that these intermediates are in fact exciplexes. A new scheme for producing free ions with a high yield using this effect and a secondary electron donor is also demonstrated. | ||||||||
 |
|
|||||||
The electron transfer (ET) quenching dynamics of excited perylene (Pe), cyanoperylene (PeCN), methanolperylene (PeOH), and methylperylene (PeMe) in N,N-dimethylaniline (DMA) has been investigated using ultrafast fluorescence up-conversion. Measurements of the rotational dynamics of PeCN and PeMe in nonpolar and polar inert solvents using optically heterodyned polarization spectroscopy are also presented. The fluorescence decay in DMA is strongly nonexponential and about 10 times faster with PeCN than with the other electron acceptors. The quenching dynamics has been analyzed with a model distinguishing three types of donor molecules surrounding the acceptor: those with optimal orientation for ET and those requiring orientational or translational diffusion prior to ET. According to this model, which can account for the whole fluorescence decay, the faster quenching dynamics of PeCN is not due to a larger ET rate constant, but to a larger number of donor molecules, typically three to four, with an optimal orientation. This is explained by the effect of dipole−dipole interaction between PeCN and the donor molecules, which favors mutual orientations with a large electronic coupling. With the other acceptors, this interaction is either not present or does not lead to ET active geometries. The occurrence of this interaction is substantiated by the rotational dynamics measurements. | ||||||||
|
||||||||
Ultrafast time-resolved fluorescence measurements have been carried out to investigate vibrational relaxation dynamics of perylene derivatives in solution. The early results obtained with cyanoperylene in acetonitrile are presented and discussed. | ||||||||
|
||||||||
The excitation of a charge transfer band by a laser pulse of finite duration and the ensuing charge recombination are calculated in the framework of the perturbation theory. The influence of the spectral characteristics of the laser pulse on the charge recombination dynamics is investigated for models including several nuclear modes that differ greatly in their timescales. It is shown that, in the area of applicability of the perturbation theory, the variation of the pulse carrier frequency inside the absorption band can significantly change the effective charge recombination rate constant. | ||||||||
|
||||||||
A transient grating setup with evanescent wave probing has been developed to investigate ultrafast processes at liquid–liquid interfaces. In order to evaluate the selectivity of this method to the interface, the speed of sound in the low refractive index medium has been measured as a function of the penetration depth of the probe pulse. Our preliminary results indicate an increase of the speed of sound in methanol with decreasing the probe depth from 100 to 70 nm. However, no correlation was found in acetonitrile in the same range. Modifications of the experiment for improving the selectivity to the interface are proposed. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel