Unpuzzle the mechanisms involved in a genetic muscle disease
Myotonic dystrophy type 1 (DM1) is the most common form of muscular dystrophy in adults, causing progressive muscle alteration and an inability to relax muscles at will. The genetic cause of the disease impairs the expression (i.e. mis-splicing) of dozens of proteins. This makes it difficult for researchers to decipher molecular changes at play in muscle deterioration.
The alteration of the connections between nerves and muscles in DM1…
In their recent publication in Skeletal Muscle, researchers from the laboratory of Prof. Perrine Castets aimed at determining whether neuromuscular junctions (NMJs) - the essential connection points between nerves and muscles – are altered in DM1. As part of the PhD project of Denis Falcetta, they showed that NMJs become fragmented before the onset of muscle degeneration, in two different mouse models of DM1. The normal pretzel shape of NMJs (left image from the Figure below) was replaced by an abnormal fragmented shape (right image from the Figure below).
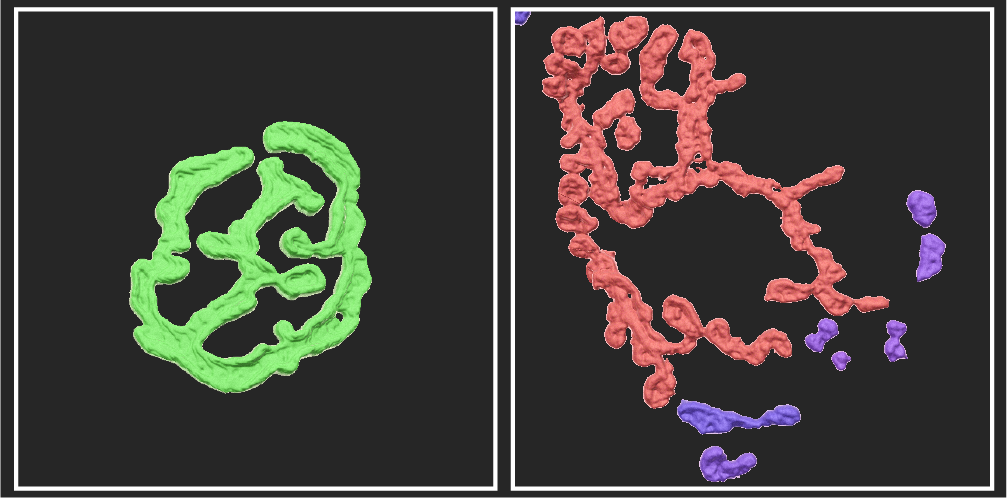 In mice with DM1, the normal pretzel shape of NMJs (left image) is replaced by an abnormal fragmented organisation (right image). Adapted from Figure S6 in Falcetta et al. 2024.
In mice with DM1, the normal pretzel shape of NMJs (left image) is replaced by an abnormal fragmented organisation (right image). Adapted from Figure S6 in Falcetta et al. 2024.
… is due to the mis-splicing of specific proteins
As these NMJ alterations may in turn contribute to muscle decline in DM1 patients, the research team set out to understand the underlying mechanisms. They discovered that the mis-splicing of specific genes, encoding Ca2+/Calmodulin-dependent kinases II, led to the deregulation of a wide set of genes involved in the maintenance of NMJs, as well as to the loss of their pretzel shape.
These findings bring one new key piece of understanding to the puzzle of muscle dysfunction in DM1. This research paves the way to the identification of new treatments that stabilize NMJs and improve muscle function for affected people.