

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
||||||||
Parent, unsubstituted porphycene and its two derivatives: 2,7,12,17-tetra-n-propylporphycene and 2,7,12,17-tetra-t-butylporphycene were substituted at the meso position with amino and nitro groups. These two families of porphycenes were characterized in detail with respect to their spectral, photophysical, and tautomeric properties. Two trans tautomers of similar energies coexist in the ground electronic state, but only one form dominates in the lowest excited singlet state. Absorption, magnetic circular dichroism (MCD), and emission anisotropy combined with quantum-chemical calculations led to the assignment of S1 and S2 transitions in both tautomers. Compared with the parent porphycene, the S1–S2 energy gap significantly increases; for one tautomeric form, the effect is twice as large as for the other. Both amino- and nitroporphycenes emit single fluorescence; previously reported dual emission of aminoporphycenes is attributed to a degradation product. Introduction of bulky t-butyl groups leads to a huge decrease in fluorescence intensity; this effect, arising from the interaction of the meso substituent with the adjacent t-butyl moiety, is particularly strong in the nitro derivative. | ||||||||



|
 |
|||||||
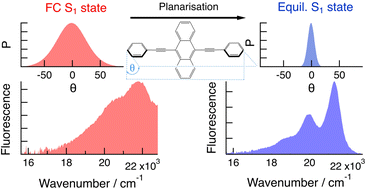
Conjugated molecules with phenylethynyl building blocks are usually characterised by torsional disorder at room temperature. They are much more rigid in the electronic excited state due to conjugation. As a consequence, the electronic absorption and emission spectra do not present a mirror-image relationship. Here, we investigate how torsional disorder affects the excited state dynamics of 9,10-bis(phenylethynyl)anthracene in solvents of different viscosities and in polymers, using both stationary and ultrafast electronic spectroscopies. Temperature-dependent measurements reveal inhomogeneous broadening of the absorption spectrum at room temperature. This is confirmed by ultrafast spectroscopic measurements at different excitation wavelengths. Red-edge irradiation excites planar molecules that return to the ground state without significant structural dynamics. In this case, however, re-equilibration of the torsional disorder in the ground state can be observed. Higher-energy irradiation excites torsionally disordered molecules, which then planarise, leading to important spectral dynamics. The latter is found to occur partially via viscosity-independent inertial motion, whereas it is purely diffusive in the ground state. This dissimilarity is explained in terms of the steepness of the potential along the torsional coordinate. | ||||||||
 |
|
|||||||
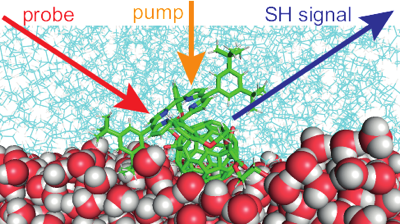
The excited-state properties of an amphiphilic porphyrin-fullerene dyad and of its porphyrin analogue adsorbed at the dodecane/water interface are investigated by using surface second-harmonic generation. Although the porphyrin is formally centrosymmetric, the second-harmonic spectra of both compounds are dominated by the intense Soret band of the porphyrin. Polarization-selective measurements and molecular dynamics simulations suggest an angle of about 45° between the donor-acceptor axis and the interfacial plane, with the porphyrin interacting mostly with the nonpolar phase. Time-resolved measurements reveal a marked concentration dependence of the dynamics of both compounds upon Q-band excitation, indicating the occurrence of intermolecular quenching processes. The significant differences in dynamics and spectra between the dyad and the porphyrin analogue are explained by a self-quenching of the excited dyad via an intermolecular electron transfer. | ||||||||
|
||||||||
A series of nine soluble, symmetric chalcogenophenes bearing hexyl-substituted triphenylamines, indolocarbazoles, or phenylcarbazoles was designed and synthesized as potential two-photon absorption (2PA) initiators. A detailed photophysical analysis of these molecules revealed good 2PA properties of the series and, in particular, a strong influence of selenium on the 2PA cross sections, rendering these materials especially promising new 2PA photoinitiators. Structuring and threshold tests proved the efficiency and broad spectral versatility of two selenium-containing lead compounds as well as their applicability in an acrylate resin formulation. A comparison with commercial photoinitiators Irg369 and BAPO as well as sensitizer ITX showed that the newly designed selenium-based materials TPA-S and TPA-BBS outperform these traditional initiators by far both in terms of reactivity and dose. Moreover, by increasing the ultralow concentration of TPA-BBS, a further reduction of the polymerization threshold can be achieved, revealing the great potential of this series for application in two-photon polymerization (2PP) systems where only low laser power is available. | ||||||||
|
 |
|||||||
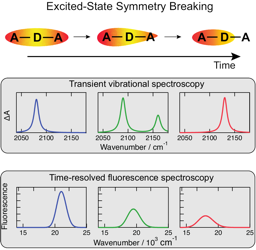
he emissive properties of symmetric molecules containing several donor-acceptor branches are often similar to those of the single-branched analogues. This is due to the at least partial localization of the excitation on one branch. Detailed understanding of this excited-state symmetry breaking (ES-SB) requires the ability to monitor this process in real time. Over the past few years, several spectroscopic approaches were shown to enable visualization of ES-SB and of its dynamics. They include the detection of new vibrational or electronic absorption bands associated with transitions that are forbidden in the symmetric excited state. Alternatively, ES-SB can be detected by observing transitions that become weaker or vanish upon localization of the excitation. Herein, we discuss these different approaches as well as their merits and weaknesses. | ||||||||
 |
|
|||||||
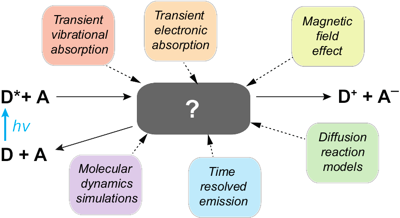
The current developments in photoredox chemistry are stimulating a renewed interest for bimolecular photoinduced electron transfer reactions. Their investigation, initiated in the 1960s using conventional photochemical tools, resulted in a relatively simple reaction scheme. More recent studies, using not only spectroscopic techniques with better time resolution and extended spectral/temporal windows but also molecular dynamics simulations, reveal a more complex picture. This Perspective focuses on the results of these latest studies with neutral organic reactants, highlighting the time dependence of the quenching rate, the effect of mutual orientation of the reactants on the electronic coupling, and their consequence on the nature of the reaction product. Remaining questions, such as the occurrence of distant electron transfer in nonviscous liquids are also addressed, and possible directions toward their answer are proposed. | ||||||||
|
 |
|||||||
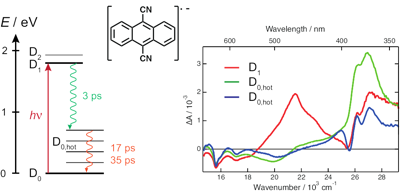
The radical anion of 9,10-dicyanoanthracene (DCA) has been suggested to be a promising chromophore for photoredox chemistry, due to its nanosecond excited-state lifetime determined from indirect measurements. Here, we investigate the excited-state dynamics of the radical anion of three cyanoanthracenes, including DCA˙−, produced by photoinduced electron transfer in liquid using both pump–probe and pump–pump probe transient electronic absorption spectroscopy. All three excited radical ions are characterised by a 3–5 ps lifetime, due to efficient non-radiative deactivation to the ground state. The decay pathway most probably involves D1/D0 conical intersection(s), whose presence is favoured by the enhanced flexibility of the radical anions relative to their neutral counterparts. The origin of the discrepancy with the nanosecond lifetime of DCA˙−* reported previously is discussed. These very short lifetimes limit, but do not preclude, photochemical applications of the cyanoanthracene anions. | ||||||||

Download this list in a RIS file or a BIB file or a PDF file
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel