Research
Ultrafast two-dimensional infrared spectroscopy (2D-IR) is our group's main workhorse. This technique allows us to resolve vibrational couplings, orientation, population exchange and energy transfer, and solvation dynamics (through the observed lineshapes in the spectra).
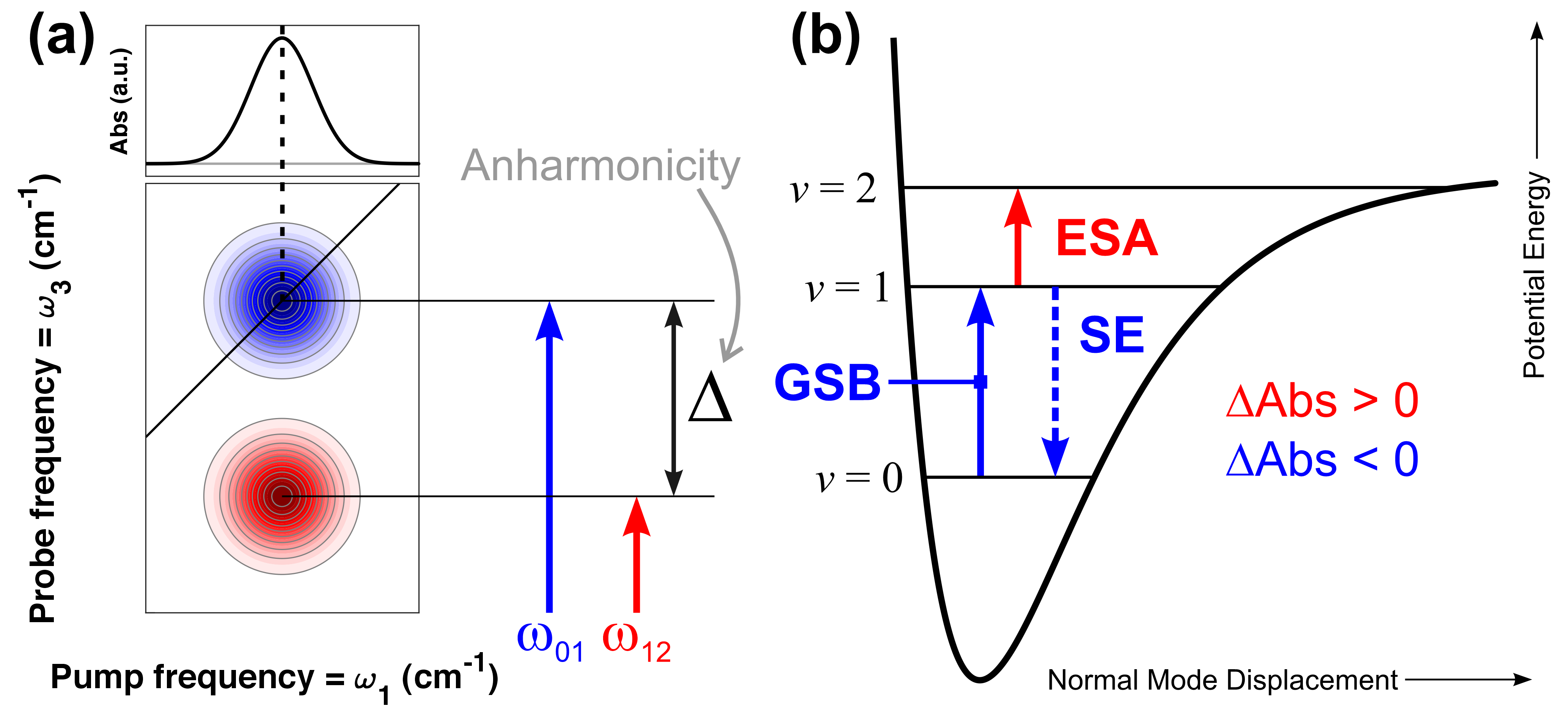
In a 2D-IR spectrum (a), every diagonal peak contains two contributions: a negative transient absorption signal due to the GSB/SE of the 0-1 transition; and a positive, anharmonically shifted ESA, due to the 1-2 transition. In conventional potential energy surfaces (b), the anharmonicity is positive, and thus the ESA is shifted to lower frequencies (wavenumbers). Cross peaks indicate vibrational couplings and/or population transfer, depending on their kinetics (analogous to COSY and NOESY cross peaks in 2D-NMR).
Transient 2D-IR (tr2DIR) is an extension of 2D-IR spectroscopy, where we now record a 2D-IR spectrum of the excited-state species or photoproducts that are generated with an actinic UV/Vis pulse.
Thanks to pulse shaping, we can record 2DIR/tr2DIR spectra in both time and frequency domains. Each approach has its own (dis)advantages. Furthermore, we can measure in the rotating frame and use t1 undersampling to greatly increase acquisition speed. [Read more in the Tutorials section]
Transition Metal Hydrides are ubiquitous intermediates in many chemical, photochemical and electrochemical processes, playing a key role in water/proton reduction catalysis and CO2 reduction.
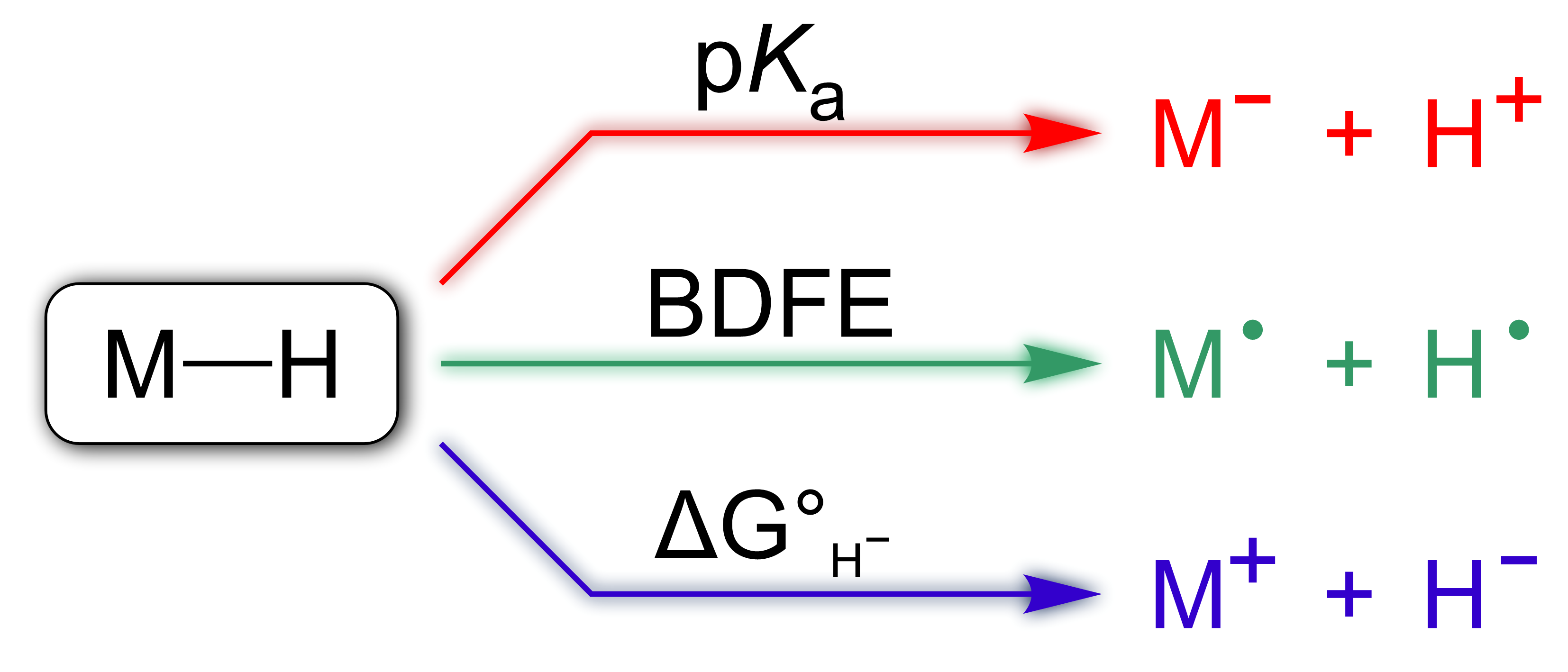 Metal hydrides can be formed (or can react) by three main pathways, controlled by the pKa, bond dissociation free energy (BDFE), and hydricity, respectively (see figure). The relative magnitudes of these three parameters depend strongly on the ability of the solvent to accommodate changes in the charge and/or electron density in the metal complex, as well as its capacity to solvate H+/·/-.
Metal hydrides can be formed (or can react) by three main pathways, controlled by the pKa, bond dissociation free energy (BDFE), and hydricity, respectively (see figure). The relative magnitudes of these three parameters depend strongly on the ability of the solvent to accommodate changes in the charge and/or electron density in the metal complex, as well as its capacity to solvate H+/·/-.
The M-H moiety has other interesting properties: its ambiguous polarisation, its ability to form hydrogen bonds, and—more importantly for us—the ultrafast dynamics of the M-H stretching vibrational mode(s). These modes directly report (thanks to 2D-IR spectroscopy) on the local field fluctuations around the M-H moiety, and can be used to determine the oxidation and protonation state of a metal hydride that can engage in PCET and/or PT/ET. In our group, we take advantage of these properties to monitor such reactions in real time.