Research
Recently, we demonstrated the influence of solvent relaxation on ESPT from a 1,8-naphthalimide-based photoacid to solvent (DMSO). The reaction dynamics were investigated using time-resolved broadband spectroscopies. The fluorescence spectra (Figure 3A) were particularly informative and allowed us to resolve all excited-state species both spectrally and kinetically. We analyzed the spectra using a newly developed global analysis based on the two-step Eigen-Weller model. The spectra together with the analysis is schematically demonstrated in Figure 3. Each of the excited-state species (ROH*, CIP*, RO–*) have spectrally distinct fluorescence bands, which in the analysis are represented by time-dependent log-normal functions (LNFs). The dynamic solvent relaxation is explicitly accounted by allowing the LNFs to redshift in frequency. Moreover, the areas of the LNFs are proportional to the concentrations and are modeled according to the rate equations derived from the Eigen-Weller model (Figure 3C, top). This allowed us to follow the fluorescence band positions (Figure 3B) and relative concentrations (Figure 3C, bottom) of each species as a function of time. Our study showed that the initial short-range proton transfer is reversible resulting in an excited-state equilibrium between ROH* and CIP* followed by diffusion-controlled separation in to free ions (RO–*). However, close inspection of the early dynamics suggested that the relative populations of the protonated form (ROH*) and CIP*are governed by the solvent relaxation that influences the relative energies of the excited states. This constitutes a breakdown of the Eigen-Weller model, although the overall agreement between the data and the analysis using classical rate equations was excellent.

Figure 3. A) Example broadband fluorescence spectra demonstrating ESPT to solvent. The color of the spectra corresponds to the time indicated by the color bar. B) Each of the excited-state species are modeled as separate fluorescence bands, which are allowed to downshift in frequency. C) The concentrations (i.e. band areas) of the species are modeled according to the Eigen-Weller model (top). The resulting populations are shown in the bottom figure.
Besides the influence of solvent relaxation, we also focus on bimolecular ESPT from photoacids to bases in organic solvents and solvent mixtures. The work consists of determinations of the acidity constants both in the ground and excited states. The aim of the solvent mixtures is to isolate the influence of a single macroscopic solvent parameter (e.g. dielectric constant or viscosity) on the ESPT process. For example, we are using a binary solvent mixture, which has a constant viscosity but the dielectric constant depends on the composition. Secondly, the time scale of the solvent relaxation is strongly dependent on the composition. We are additionally using computational chemistry methods (DFT and TD-DFT) to gain further insight into both ground- and excited-state properties of photoacids.
Excited-state proton transfer in solutions
Proton transfer is undoubtedly one of the most significant reactions in chemical and biological processes. This reaction involves the transfer of a proton from a proton donor (AH) to a proton acceptor (B), which can be either an organic base or solvent. In most cases proton transfer results in a formation of charged products (ion pairs) and is therefore strongly influenced by the solvent environment.

Experimental investigations of proton-transfer reactions of ground-state species is complicated by the dynamic nature of the dissociation-association equilibrium. Discovery of excited-state acids, also known as photoacids, has enabled detailed time-resolved studies on the mechanism and kinetics of the dissociation process. Dissociation of the proton can be initiated by a short laser pulse and the subsequent cascade of events can be spectroscopically followed in time. Excited-state proton transfer (ESPT) serves as a model system for studying the microscopic reaction mechanism and is therefore of great fundamental interest.
Photoacids are often hydroxy-substituted aromatic compounds, which increase their acidity by up to ten orders of magnitude upon excitation to a higher electronic state. The photoacidity is generally attributed to a redistribution of the electron density (intramolecular charge-transfer) from the hydroxyl oxygen to the aromatic system upon excitation. The electronic redistribution can be seen in the frontier orbitals corresponding to the ground and excited states (Figure 1, left). This weakens the O–H bond and results in dissociation of the hydroxyl proton in the presence of a suitable proton acceptor (Figure 1, right). Due to the intramolecular charge-transfer (ICT) property, the acidity can be enhanced by introducing electron-withdrawing groups on the aromatic system. This approach has resulted in development of even stronger photoacids, also known as “super” photoacids. These compounds are characterized by negative excited-state pKa values and ability to deprotonate in alcohols and other organic solvents such as DMSO and formamides. Hence these compounds have proven to be extremely useful in investigations on ESPT in non-aqueous media where the normal photoacids do not undergo ESPT. In our research, we utilize “super” photoacids based on 1,8-naphthalimides, hydroxypyrenes, and hydroxyquinolines.

Figure 1. (left) Frontier orbitals of a 1,8-naphthalimide-based “super” photoacid. The electron density is transferred from the hydroxyl oxygens to the aromatic system upon excitation from the HOMO to the LUMO. (right) The charge shift weakens the O–H bond and the photoacid undergoes excited-state proton transfer in the presence of a suitable acceptor such as an organic base (imidazole).

After more than 60 years of research, ESPT in aqueous solutions is fairly well understood. The mechanism of the dissociation process is generally explained according to the Eigen-Weller model (Figure 3C, top), which consists of an initial short-range proton-transfer step producing contact ion pairs followed by a diffusion-controlled separation into free ions. The intermediate contact ion pairs have been experimentally observed in water and other protic solvents for strong photoacids. Moreover, detailed diffusional models accounting for the reversibility of the reaction has been successfully applied to ESPT in aqueous solutions and alcohols.
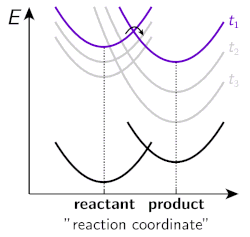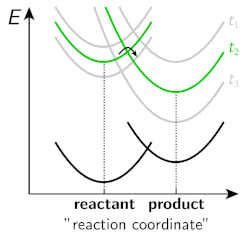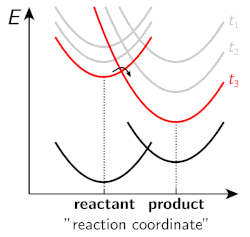
Figure 2. Energy surfaces of a “neutral” reactant and a “charged” product at different stages of solvent relaxation.
However, several fundamental aspects of ESPT in organic solvents have not been investigated in detail and our research aims to bridge that gap. One of the key research questions in our group is to investigate the influence of solvation dynamics on the rate and yield of ESPT reactions. A typical proton transfer starts from a neutral reactant, proceeds via strongly dipolar transition state and results in formation of charged products. Therefore, both the reaction barrier and the overall driving force of the reaction are strongly influenced by solvent polarity. Moreover, ESPT reactions of strong photoacids are often ultrafast (from a few picoseconds down to sub-picosecond) occurring on the same time scale with the dynamic solvent relaxation. Hence the energy landscape of the reaction changes during the initial solvent relaxation resulting in non-equilibrium reaction dynamics. This is schematically illustrated in Figure 2, which shows the energy surfaces of a “neutral” reactant and a “charged” product upon increasing time. As is evident from the figure, the reaction barrier is decreased and the driving force increased as the solvent relaxation proceeds.