Publication 5
- Computational and synthetic studies on the cyclometallation reaction of dimethylbenzylamine with [IrCl2Cp*]2: role of the chelating base
Boutadla, Y.; Davies, D. L.; Macgregor, S. A.; Poblador-Bahamonde, A. I.
Dalton Trans. 2009, 5887-5893
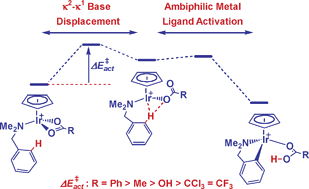
The results of a joint computational and experimental study of the cyclometallation reactions of dimethylbenzylamine (DMBA-H) with [IrCl2Cp*]2 and a range of chelating bases are presented. With acetate, density functional theory calculations on the key intermediate, [Ir(DMBA-H)(κ2-OAc)Cp]+, define a two-step C–H activation process involving initial κ2–κ1 displacement of base to give an intermediate that is stabilized by internal H-bonding. Facile C–H bond cleavage then occurs via ‘ambiphilic metal ligand activation’ (AMLA). A similar pattern is computed for other carboxylates and bicarbonate, and in each case the ease of C–H activation is governed by the accessibility of the κ2–κ1 base displacement step; thus, more weakly coordinating bases promote C–H activation. For triflate, [Ir(DMBA-H)(κ1-CF3SO3)Cp]+ is more stable than its κ2-isomer and C–H activation proceeds with a barrier of only 3.8 kcal mol−1. Experimental studies confirm that a range of carboxylates and triflate can effect cyclometallation; however, reactivity patterns are not consistent with the computed C–H activation barriers. Instead, the role of base in opening the [IrCl2Cp*]2 dimer and subsequent formation of the [Ir(DMBA-H)(base)Cp*]+ intermediates appears crucial. Calculations indicate these processes are far more favourable for acetate than for triflate.
DOI : 10.1039/b905469c
archive ouverte unige:25968