
|
Photophysics and Photochemistry of Transition Metal Compounds |
| Home Research Members Collaborations Publications |

Stay informed of our last publications with our atom feed 
Author list

2017
2016
2015
2014
2013
2012
2011
2010
2009
2008
2007
2006
2005
2004
2003
2002
2001
2000
1999
1998
1997
1996
1995
1994
1993
1992
1991
1990
1988
1986
1985
|
||||||||
The nucleation and growth properties of domains of molecules of the same state in open boundary three-dimensional (3D) spin-crossover systems of various shapes are discussed within the framework of the mechanoelastic model. The molecules are situated on face-centered-cubic lattices and are linked by springs through which they interact. Monte Carlo simulations imply that clusters nucleate from corners in the case of systems having well-developed faces and from kinks in the case of spherical samples, in accordance with available experimental data. In addition, a method to characterize the cooperativity in these systems is proposed, which by scanning the fluctuations in the 3D samples can be related directly to powder x-ray-diffraction experiments. | ||||||||
|
 |
|||||||
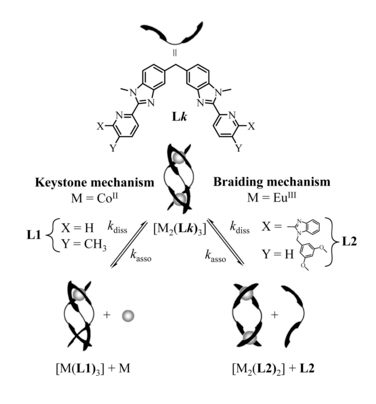
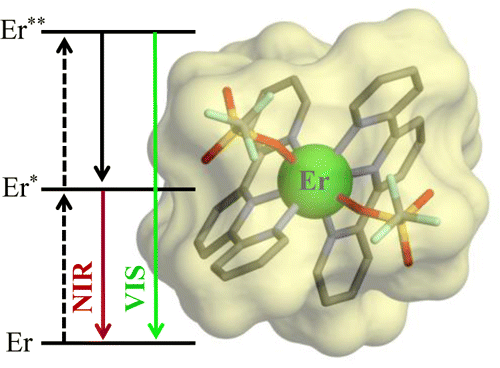
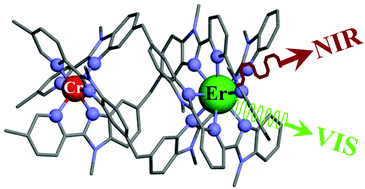
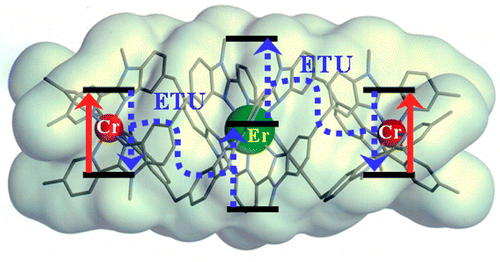
The kinetic lability of hexadentate gallium-based tripods is sufficient to ensure thermodynamic self-assembly of luminescent heterodimetallic [GaLn(L3)3]6+ helicates on the hour time scale, where Ln is a trivalent 4f-block cation. The inertness is however large enough for preserving the triple-helical structure when [GaLn(L3)3]6+ is exposed to lanthanide exchange. The connection of a second gallium-based tripod further slows down the exchange processes to such an extent that spectroscopically active [CrErCr(L4)3]9+ can be diluted into closed-shell [GaYGa(L4)3]9+ matrices without metal scrambling. This feature is exploited for pushing molecular-based energy transfer upconversion (ETU) at room temperature. | ||||||||
 |
|
|||||||
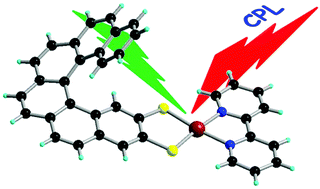
Chiral metal dithiolene complexes represent a family of chiral precursors, which can give rise to molecular materials with properties resulting from the interplay of chirality with conductivity, magnetism, and photophysics. We describe herein the first examples of chiral metal diimine dithiolene complexes, by the use of a platinum(II) centre coordinated by 2,2’-bipyridine and helicene-dithiolene ligands. Straightforward synthesis of racemic and enantiopure complexes allows the preparation of luminescent Pt(bipy) [4] and [6]helicene compounds for which the solid-state structure was determined as well. TD-DFT calculations support the assignment of the low energy bands observed in the UV-vis absorption spectra as mixed metal-ligand-to-ligand charge transfer transitions and confirm that the emission band results from the T1 excited state. Interestingly the enantiopure [6]helicene complexes show CPL activity at room temperature in acetonitrile solutions with anisotropy factors of 3×10-4. | ||||||||

|
 |
|||||||
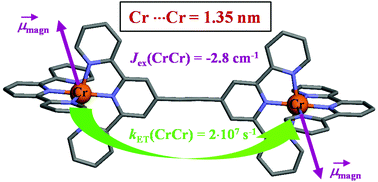
Compared with divalent ruthenium coordination complexes, which are widely exploited as parts of multi-component photonic devices, optically active trivalent chromium complexes are under-represented in multi-metallic supramolecular architectures performing energy conversion because of the tricky preparation of stable heteroleptic CrIII building blocks. We herein propose a kind of remedy with the synthesis of a novel family of kinetically inert hetereloptic bis-terdentate mononuclear complexes, which can be incorporated into dinuclear rod-like diads as a proof-of-concept. The mechanism and magnitude of intermetallic Cr···Cr communications have been unraveled by a combination of magnetic, photophysical and thermodynamic investigations. Alternated aromatic/alkyne connectors provided by Sonogashira coupling reactions emerge as the most efficient wires for long-distance communication between two chromium centres bridged by Janus-type back-to-back bis-terdentate receptors. | ||||||||
 |
|
|||||||
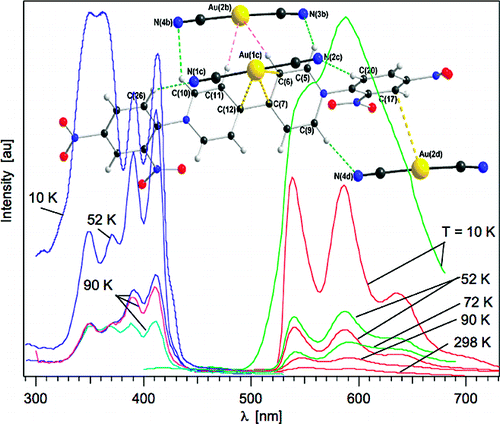
K3Fe(CN)6 reacts with the viologen 1,1′-bis(2,4-dinitrophenyl)-4,4′-bipyridinium dication, (DNP)2+, to form a supramolecular complex, (DNP)3[Fe(CN)6]2·10H2O (1). The crystal structure of 1 reveals that there are two [Fe(CN)6]3– anions within an organic framework of three (DNP)2+ cations with the shortest Fe(III)···Fe(III) distances of ca. 9.8 Å, distances that minimize extensive long-range magnetic exchange coupling interactions between the [Fe(CN)6]3– anions, and, thus, 1 is paramagnetic above ca. 17 K and exhibits weak ferromagnetic coupling between 17 and 3 K and antiferromagnetic coupling between 3 and 1.8 K. The long Fe(III)···Fe(III) distances permit slow spin–spin and slow spin–lattice paramagnetic relaxation, relative to the iron-57 Larmor precession frequency, as is evidenced by the Mössbauer spectra measured between 3 and 60 K; between 85 and 295 K, rapid paramagnetic relaxation is observed. Both the slow spin–spin and slow spin–lattice relaxation are mediated by the organic, π-conjugated viologen cations. The Fe–C distances, the Mössbauer isomer shifts, the temperature dependence of the magnetic susceptibility, and the 3 K magnetization results all indicate the presence of low-spin Fe(III) ions in the [Fe(CN)6]3– anions in 1. There is no unequivocal indication of the presence of any formal electron delocalization or transfer from the [Fe(CN)6]3– anion to the (DNP)2+ cations in the results obtained from X-ray crystallography, magnetic measurements, and Mössbauer spectra. Because of enhancement of the spin–orbit coupling by the heavy-atom or -ion effect, the Fe(III) ions in the [Fe(CN)6]3– anions interact with the (DNP)2+ cations, causing them to fluoresce with increasing intensity upon cooling from 90 to 25 K when excited at 300 nm. The resulting luminescence of the viologen (DNP)2+ cation induced by the [Fe(CN)6]3– anions indicates the presence of significant mixing of the molecular orbitals derived from the [Fe(CN)6]3– anions and the molecular orbitals associated with the (DNP)2+ cations to yield bonding supramolecular orbitals in 1, a mixing that is also observed between 50 and 3 K in the temperature dependence of the isomer shift of 1. | ||||||||
|
 |
|||||||
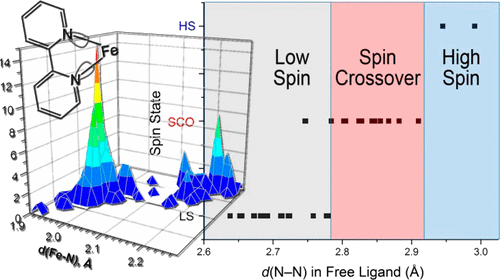
We propose a simple method for predicting the spin state of homoleptic complexes of the Fe(II) d6 ion with chelating diimine ligands. The approach is based on the analysis of a single metric parameter within a free (noncoordinated) ligand: the interatomic separation between the N-donor metal-binding sites. An extensive analysis of existing complexes allows the determination of critical N···N distances that dictate the regions of stability for the high-spin and low-spin complexes, as well as the intermediate range in which the magnetic bistability (spin crossover) can be observed. The prediction has been tested on several complexes that demonstrate the validity of our method. | ||||||||
|
||||||||
Introduction of heterocycles in the helical skeleton of helicenes allows modulation of their redox, chiroptical and photophysical properties. Herein, we describe the straightforward preparation and structural characterization by single crystal X-ray diffraction of thiadiazole-[7]helicene, which has been resolved into (M) and (P) enantiomers by chiral HPLC, together with its S-shaped double [4]helicene isomer, as well as the smaller congeners thiadiazole-[5]helicene and benzothiadiazole-anthracene. A copper(II) complex with two thiadiazole-[5]helicene ligands has been structurally characterized and it shows the presence of both (M) and (P) isomers coordinated to the metal centre. The emission properties of the unprecedented heterohelicenes are highly dependent on the helical turn, as the [7]- and [5]helicene are poorly emissive, whereas their isomers, that is, the S-shaped double [4]helicene and thiadiazole-benzanthracene, are luminescent, with quantum efficiencies of 5.4% and 6.5%, respectively. DFT calculations suggest a quenching of the luminescence of enantiopure [7]helicenes through an intersystem crossing mechanism arising from the relaxed excited S1 state. | ||||||||
|
||||||||
Ultrafast time-resolved infrared spectroscopy of [Ru(bpy)3]2+ (bpy = 2,2’-bipyridine), [Ru(mbpy)3]2+ (mbpy = 6-methyl-2,2’-bipyridine), and [Ru(mphen)3]2+ (mphen = 2-methyl-1,10’-phenanthroline) in deuterated acetonitrile serves to elucidate the evolution of the system following pulsed excitation into the 1MLCT band at 400 nm. Whereas for [Ru(bpy)3]2+ no intermediate state can be evidenced for the relaxation of the corresponding 3MLCT state back to the ground state, for [Ru(mbpy)3]2+ and [Ru(mphen)3]2+ an intermediate state with a lifetime of about 400 ps is observed. The species associated IR difference spectra of this state are in good agreement with the calculated difference spectra of the lowest energy 3dd state using DFT. The calculated potential energy curves for all the complexes in the triplet manifold along the metal-ligand distance show that for [Ru(bpy)3]2+ the 3dd state is at higher energy than the 3MLCT state and that there is a substantial barrier between the two minima. For [Ru(mbpy)3]2+ and [Ru(mphen)3]2+, the 3dd state is at lower energy than the 3MLCT state. | ||||||||

 |
|
|||||||
The manganese-nitronyl-nitroxide two dimensional coordination polymer {[Mn2(NITIm)3]ClO4}n (1) (NITImH = 2-(2-imidazolyl)-4,4,5,5-tetramethyl-4,5-dihydro-1H-3-oxide-1-oxyl) undergoes an unusual hysteretic thermoinduced valence tautomeric transition near room temperature, during which the manganese(II) ions are oxidized to manganese(III) and two of the three deprotonated radicals (NITIm-) are reduced to their diamagnetic aminoxyl form (denoted NITImRed2-). Upon cooling, the high-temperature species {[MnII2(NITIm)3]ClO4}n (1HT) turns into the low-temperature species {[MnIII2(NITImRed)2(NITIm)]ClO4}n (1LT) around 274 K, while on heating the process is reversed at about 287 K. This valence tautomeric phenomenon is supported by temperature-dependent magnetic susceptibility measurements, differential scanning calorimetry (DSC), crystal structure determination, UV-vis absorption, X-ray absorption (XAS) an emission (XES) and Electron Paramagnetic Resonance (EPR) spectroscopies in the solid-state. | ||||||||
|
 |
|||||||
Here we reproduce the static and dynamical properties of spin-crossover complexes in the framework of the mechanoelastic model applied to triangular lattices. The switching processes between the high-spin and low-spin states are studied by combining the Monte Carlo method with the elastic lattice relaxation. The transition probabilities between the two states take into account intrinsic parameters, the values of which are approximated from experimental quantities (e.g., the energy gap, and the degeneracy ratio from the thermodynamic enthalpy and the entropy difference between the states), and the elastic force or elastic energy stored in the springs connecting the spin-changing centres. The value of the corresponding spring constant is estimated from the experimentally determined variation of the ligand-field strengths in the two spin states due to the cooperativity and the bulk modulus. Both simulated hysteresis loops and relaxation curves are in agreement with experimental data. Cooperativity related phenomena such as like-spin domain formation and the evolution of the interaction distribution with the HS fraction are also analysed. | ||||||||
 |
|
|||||||
Considered at the beginning of the 21th century as being incompatible with the presence of closely bound high-energy oscillators, lanthanide-centered superexcitation, which is the raising of an already excited electron to an even higher level by excited-state energy absorption, is therefore a very active topic strictly limited to the statistical doping of low-phonon bulk solids and nanoparticles. We show here that molecular lanthanide-containing coordination complexes may be judiciously tuned to overcome these limitations and to induce near-infrared (NIR)-to-visible (VIS)-light upconversion via the successive absorption of two low-energy photons using linear-optical responses. Whereas single-ion-centered excited-state absorption mechanisms remain difficult to implement in lanthanide complexes, the skillful design of intramolecular intermetallic energy-transfer processes operating in multimetallic architectures is at the origin of the recent programming of erbium-centered molecular upconversion. | ||||||||
|
||||||||
Optical spectroscopy of transition metal complexes plays an important role in establishing excited state electronic and nuclear structures and thus in the elucidation of the multitude of photophysical and photochemical relaxation processes. The most important advances in this area of research over the past decade are due to the development of new experimental techniques such as ultrafast spectroscopy as well as structure determination in conjunction with other methods such as high pressure and variable temperature techniques. In this contribution, several paradigmatic systems, namely of complexes if chromium(III), iron(II), ruthenium(II), nickel(II), platinum(II) and palladium(II), are discussed with regard to their excited electronic and nuclear structures and photophysical relaxation processes. | ||||||||
|
 |
|||||||
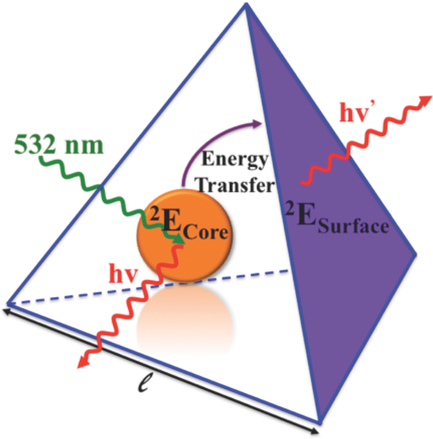
Size-controlled nanocrystals (140 nm and 670 nm) and microcrystals (2.5 mm) of the three-dimensional oxalate network [Ru(bpy)3][NaCr(ox)3], ox = oxalate, bpy = 2,2’-bipyridine, were prepared by the reverse micelle technique. The photo-physical properties of the [Cr(ox)3]3- chromophores in the nanocrystals at low temperatures are significantly different from those of the same chromophore in 4 mm crystallites prepared by fast precipitation. For the latter, the absorption in the region of the R lines of the 4A2 → 2E transition is characterized by a sharp doublet. For the nanocrystals the inhomogeneous broadening of the two lines is considerably larger with tails on the low-energy side. Whereas the 4 mm crystallites at low temperatures just show equally sharp emission from the R1 line, the emission intensity from the nanocrystallites is shifted into the low-energy tail. Time resolved fluorescence line narrowing spectra and luminescence decay curves demonstrate that this is due to efficient directional energy migration from the center of the nanocrystals towards the surface | ||||||||
|
||||||||
The location of the Pd atoms in Pd2Au36(SC2H4Ph)24, is studied both experimentally and theoretically. X-ray photoelectron spectroscopy (XPS) indicates oxidized Pd atoms. Palladium K-edge extended X-ray absorption fine-structure (EXAFS) data clearly show Pd-S bonds, which is supported by far infrared spectroscopy. By comparing theoretical EXAFS spectra in R space and circular dichroism spectra of the staple, surface and core doped structures with experimental spectra. | ||||||||
 |
|
|||||||
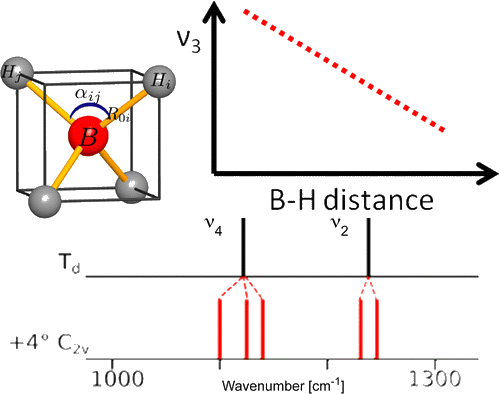
Among the different potential hydrogen storage materials, borohydrides have been largely investigated because of their high gravimetric and volumetric hydrogen content. In the analysis of borohydrides, vibrational spectroscopy plays an important role since it gives information on the local structure of the BH4– ion inside the solid. Here the GF method, developed by Wilson, is used in order to determine the local symmetry of BH4– in solid borohydrides starting from their vibrational spectra. Two different cases of deformations of BH4– are considered. In the first case, the effects of small angular variations on the vibrational spectra of borohydrides will be taken into account; starting from the splitting of the bands corresponding to the deformation modes, the angular deformations will be estimated. In the second one, the BH4– under chemical pressure (in different cubic alkali halides) is considered; in this case, the symmetry of the BH4– remains Td, while the bond lengths change according to the pressure experienced. Different practical examples will be illustrated. | ||||||||
|
 |
|||||||
The ligand 3-chloro-6-dipicolylamino-1,2,4,5-tetrazine (Cl-TTZ-dipica) 1, prepared by the direct reaction between 3,6-dichloro-1,2,4,5-tetrazine and di(2-picolyl)-amine, afforded a series of four neutral transition metal complexes formulated as [Cl-TTZ-dipica-MCl2]2, with M = Zn(II) 2a, Cd(II) 2b, Mn(II) 2c and Co(II) 2d, when reacted with the corresponding metal chlorides. The dinuclear structure of the isostructural complexes was disclosed by single crystal X-ray analysis, clearly indicating the formation of [MII-(m-Cl)2MII] motifs and the involvement of the amino nitrogen atom in semi-coordination with the metal centers, thus leading to distorted octahedral coordination geometries. Moreover, the chlorine atoms, either coordinated to the metal or as substituent on the tetrazine ring, engage respectively in specific anion-p intramolecular and intermolecular interactions with the electron poor tetrazine units in the solid state, thus controlling the supramolecular architecture. Modulation of the emission properties is observed in the case of the Zn(II) and Cd(II) complexes when compared to the free ligand. A striking difference is observed in the magnetic properties of the Mn(II) and Co(II) complexes. An antiferromagnetic coupling takes place in the dimanganese(II) compound (J = -1.25 cm-1) while the Co(II) centers are ferromagnetically coupled in the corresponding complex (J = +0.55 cm-1), the spin Hamiltonian being defined as H = -JSA.SB. | ||||||||
|
||||||||
The recently obtained spin-crossover nanoparticles are possible candidates for applications in the recording media industry as materials for data storage, or as pressure and temperature sensors. For these applications, the intermolecular interactions and interactions between spin-crossover nanoparticles are extremely important, as they may be essential factors in triggering the transition between the two stable phases: the high-spin and low-spin ones. In order to find correlations between the distributions in size and interactions and the transition temperatures distribution, we apply the FORC (First Order Reversal Curves) method, using simulations based on a mechanoelastic model applied to 2D triangular lattices composed of molecules linked by springs and embedded in a surfactant. We consider two Gaussian distributions: one is the size of the nanoparticles and another is the elastic interactions between edge spin-crossover molecules and the surfactant molecules. In order to disentangle the kinetic and non-kinetic parts of the FORC distributions, we compare the results obtained for different temperature sweeping rates. We also show that the presence of few larger particles in a distribution centered around much smaller particles dramatically increases the hysteresis width. | ||||||||
 |
|
|||||||
The synthesis, crystal structure and photophysical properties of the new compound [Mn4(ThiaSO2)2F][K(18-crown-6)], ThiaSO2 = p-tertbutylsulphonylcalix[4]arene, are presented and compared to the ones of [Mn4(ThiaSO2)2F]K. The strong orange luminescence is attributed to the Mn2+ centred 4T1 → 6A1 transition. Its temperature and pressure dependence and quenching by molecular dioxygen are reported. The latter is attributed to energy transfer from the 4T1 state exciting dioxygen to its 1Σ+g state. In the solid state, the quenching is much more efficient in [Mn4(ThiaSO2)2F][K(18-crown-6)] than in [Mn4(ThiaSO2)2F]K. This is attributed to the open pore structure of the former allowing fast diffusion of dioxygen into the crystal lattice. | ||||||||
|
 |
|||||||
Size-controlled micro- and nanocrystals of a [Ru(bpy)3][NaCr(ox)3] coordination network are prepared using reverse micelles. Compared with the bulk reference, the 2E emission of the Cr(III) ions indicates an efficient directional energy migration toward the surface of the nanocrystals. | ||||||||
 |
|
|||||||
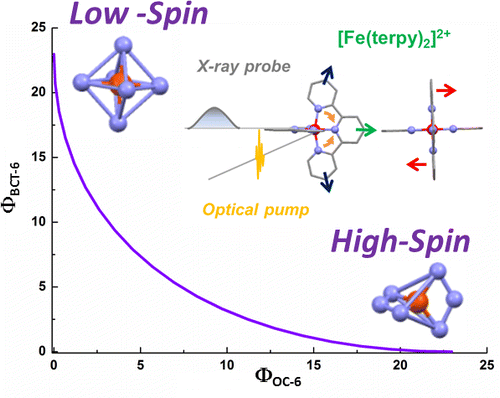
Establishing a tractable yet complete reaction coordinate for the spin-state interconversion in d4–d7 transition metal complexes is an integral aspect of controlling the dynamics that govern their functionality. For spin crossover phenomena, the limitations of a single-mode approximation that solely accounts for an isotropic increase in the metal–ligand bond length have long been recognized for all but the simple octahedral monodentate FeII compounds. However, identifying the coupled deformations that also impact on the unimolecular rate constants remains experimentally and theoretically challenging, especially for samples that do not display long-range order or when crystallization profoundly alters the dynamics. Owing to the rapid progress in ultrafast X-ray absorption spectroscopy (XAS), it is now possible to obtain transient structural information in any physical phase with unprecedented details. Using picosecond XAS and DFT modeling, the structure adopted by the photoinduced high-spin state of solvated [Fe(terpy)2]2+ (terpy: 2,2′:6′,2″-terpyridine) has been recently established. Based on these results, the methodology of the continuous shape measure is applied to classify and quantify the short-lived distortion of the first coordination shell. The reaction coordinate of the spin-state interconversion is clearly identified as a double axial bending. This finding sets a benchmark for gauging the influence of first-sphere and second-sphere interactions in the family of FeII complexes that incorporate terpy derivatives. Some implications for the optimization of related photoactive FeII complexes are also outlined. | ||||||||
|
||||||||
Characterizing structural distortions in the metastable spin states of d4–d7 transition metal ion complexes is crucial to understand the nature of their bistability and eventually control their switching dynamics. In particular, the impact of the Jahn–Teller effect needs to be assessed for any electronic configuration that could be effectively degenerate, as in e.g. the high-spin (HS) manifold of highly symmetric homoleptic FeII complexes. However, capturing its manifestations remains challenging since crystallization generally alters the molecular conformations and their interconversion. With the rapid progress of ultrafast X-ray absorption spectroscopy, it is now possible to collect data with unprecedented signal-to-noise ratio, opening up for detailed structural characterization of transient species in the homogeneous solution phase. By combining the analysis of picosecond X-ray absorption spectra with DFT simulations, the structure of the photoinduced HS state is elucidated for solvated [Fe(terpy)2]2+ (terpy = 2,2′:6′,2″-terpyridine). This species can be viewed as the average 5B structure in D2 symmetry that originates from a dynamic Jahn–Teller effect in the HS manifold. These results evidence the active role played by this particular instance of vibronic coupling in the formation of the HS state for this benchmark molecule. Ultimately, correlating the interplay between intramolecular and intermolecular degrees of freedom to conformational strain and distortions in real time should contribute to the development of advanced functionalities in transition metal ion complexes. | ||||||||
|
 |
|||||||
Bis(thiomethyl)- and bis(thiohexyl)-tetrathiafulvalene-bromo-benzothiadiazoles, containing electron donor tetrathiafulvalene (TTF) and electron acceptor benzothiadiazole (BTD) units, have been prepared by Stille coupling reactions between the TTF-SnMe3 precursors and BTD-Br2. In another series of experiments, TTF-acetylene-BTD compounds have been synthesized by Sonogashira coupling between either TTF-acetylenes and BTD-Br2 in low yields, or TTF-iodine and BTD-acetylene in moderate yields. In the compound TTF-C≡C-BTD the TTF and BTD units are coplanar in the solid state, as shown by the single crystal X-ray structure, and there is segregation in the packing between the donor and acceptor units. All the derivatives have good electron donor properties, as determined by cyclic voltammetry measurements, and they can also be reversibly reduced thanks to the presence of the BTD moiety. UV-visible spectroscopy and photophysical investigations show the presence of an intramolecular charge transfer (ICT) band and an emission band originating from the charge transfer. Both the absorption and the emission are modulated by the substitution scheme and the insertion of the acetylenic bridge. | ||||||||
 |
|
|||||||
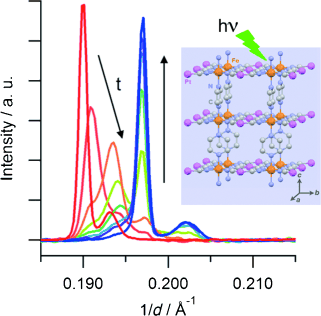
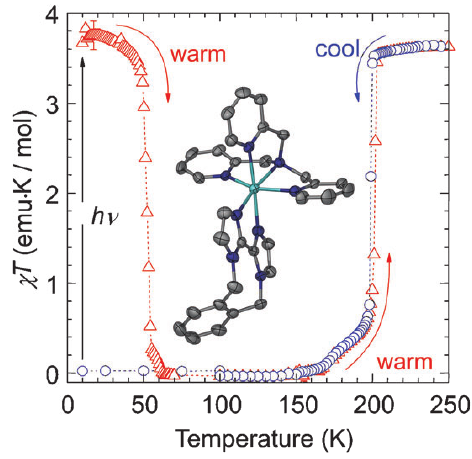
The Hoffman-type coordination compound [Fe(pz)Pt(CN)4]⋅2.6 H2O (pz=pyrazine) shows a cooperative thermal spin transition at around 270 K. Synchrotron powder X-Ray diffraction studies reveal that a quantitative photoinduced conversion from the low-spin (LS) state into the high-spin (HS) state, based on the light-induced excited spin-state trapping effect, can be achieved at 10 K in a microcrystalline powder. Time-resolved measurements evidence that the HS→LS relaxation proceeds by a two-step mechanism: a random HS→LS conversion at the beginning of the relaxation is followed by a nucleation and growth process, which proceeds until a quantitative HS→LS transformation has been reached. | ||||||||
|
 |
|||||||
This work shows that the operation of near-infrared to visible light-upconversion in a discrete molecule is not limited to non-linear optical processes, but may result from superexcitation processes using linear optics. The design of nine-coordinate metallic sites made up of neutral N-heterocyclic donor atoms in kinetically inert dinuclear [GaEr(L1)3]6+ and trinuclear [GaErGa(L2)3]9+ helicates leads to [ErN9] chromophores displaying unprecedented dual visible nanosecond Er(4S3/2→4I15/2) and near-infrared microsecond Er(4I13/2→4I15/2) emissive components. Attempts to induce one ion excited-state absorption (ESA) upconversion upon near-infrared excitation of these complexes failed because of the too-faint Er-centred absorption cross sections. The replacement of the trivalent gallium cation with a photophysically-tailored pseudo-octahedral [CrN6] chromophore working as a sensitizer for trivalent erbium in [CrEr(L1)3]6+ improves the near-infrared excitation efficiency, leading to the observation of a weak energy transfer upconversion (ETU). The connection of a second sensitizer in [CrErCr(L2)3]9+ generates a novel mechanism for upconversion, in which the superexcitation process is based on the CrIII-sensitizers. Two successive Cr→Er energy transfer processes (concerted-ETU) compete with a standard Er-centred ETU, and a gain in upconverted luminescence by a factor larger than statistical values is predicted and observed. | ||||||||
|
||||||||
The role of ligand-field states for the photophysical properties of d6 systems has been discussed in a large number of publications over the past decades. Since the seminal paper by Houten and Watts, for instance, the quenching of the 3MLCT luminescence in ruthenium(II) polypyridyl complexes is attributed to the presence of the first excited ligand-field state, namely a component of the 3T1(t2g5eg1) state, at similar energies. If this state lies above the 3MLCT state, the luminescence is quenched via thermal population at elevated temperatures only. If it lies well below, then the luminescence is quenched down to cryogenic temperatures. In this contribution we present transient absorption spectra on non-luminescent ruthenium polypyridyl complexes such as [Ru(m-bpy)3]2+, m-bpy = 6-methyl-2,2’-bipyridine, in acetonitrile at room temperature, which reveal an ultra-rapid depopulation of the 3MLCT state but a much slower ground state recovery. We propose that in this and related complexes the methyl groups force longer metal-ligand bond lengths, thus resulting in a lowering of the ligand-field strength such that the 3dd state drops to below the 3MLCT state, and that furthermore the population of this state from the 3MLCT state occurs faster than its decay to the ground state. In addition we demonstrate that in this complex the luminescence can be switched on by external pressure, which we attribute to a destabilisation of the ligand-field state by the pressure due to its larger molecular volume compared to the ground state as well as the 3MLCT state. | ||||||||
 |
|
|||||||
Perovskite materials host an incredible variety of functionalities. Although the lightest element, hydrogen, is rarely encountered in oxide perovskite lattices, it was recently observed as the hydride anion H−, substituting for the oxide anion in BaTiO3. Here we present a series of 30 new complex hydride perovskite-type materials, based on the non-spherical tetrahydroborate anion BH4− and new synthesis protocols involving rare-earth elements. Photophysical, electronic and hydrogen storage properties are discussed, along with counterintuitive trends in structural behaviour. The electronic structure is investigated theoretically with density functional theory solid-state calculations. BH4-specific anion dynamics are introduced to perovskites, mediating mechanisms that freeze lattice instabilities and generate supercells of up to 16 × the unit cell volume in AB(BH4)3. In this view, homopolar hydridic di-hydrogen contacts arise as a potential tool with which to tailor crystal symmetries, thus merging concepts of molecular chemistry with ceramic-like host lattices. Furthermore, anion mixing BH4−←X− (X−=Cl−, Br−, I−) provides a link to the known ABX3 halides. | ||||||||
|
 |
|||||||
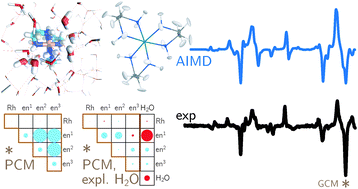
Backscattered Raman optical activity (ROA) spectra are measured for Δ- and Λ-tris-(ethylenediamine)rhodium(III) chloride in aqueous solution. In addition, the spectra of the four possible conformers in the Λ configuration are investigated by ab initio calculations. The Λ(δδδ) conformer is in best agreement with experimental spectra and examined in more details. The two most stable conformers according to the calculations are not compatible with the experimental ROA spectrum. Insights into the origin of observed band intensities are obtained by means of group coupling matrices. The influence of the first solvation shell is explored via an ab initio molecular dynamics simulation. Taking explicit solvent molecules into account further improves the agreement between calculation and experiment. Analysis of selected normal modes using group coupling matrices shows that solvent molecules lead to normal mode rotation and thus contribute to the ROA intensity, whereas the contribution of the Rh can be neglected. | ||||||||
 |
|
|||||||
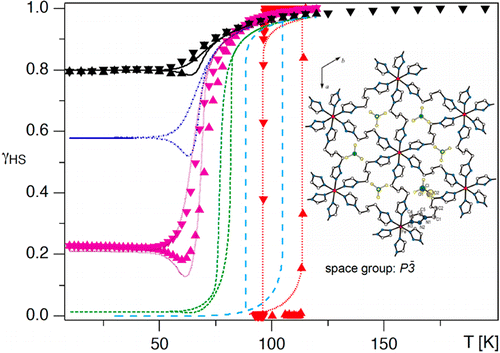
Depending on the iron(II) concentration, the mixed crystals of {[Zn1-xFex(bbtr)3](BF4)2}∞, bbtr = 1,4-di(1,2,3-triazol-1-yl)butane, 0.01 ≤ x ≤ 1, show macroscopic light-induced bistability between the high-spin and the low-spin state. In the highly diluted system with x = 0.01 and up to x = 0.31, the photoinduced low-spin state always relaxes back to the high-spin state independent of the initial light-induced low-spin fraction. In the highly concentrated mixed crystals with x = 0.67, 0.87 and 1, the strong cooperative effects coupled to a crystallographic phase transition result in light-induced bistability with decreasing critical light-induced low-spin fraction and increasing hysteresis width for increasing iron(II) concentrations. The lower limit for the light-induced bistability is estimated at x ≈ 0.5. | ||||||||
|
 |
|||||||
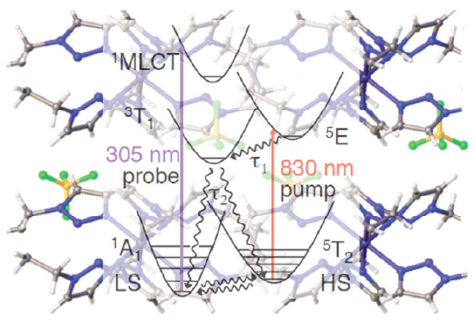
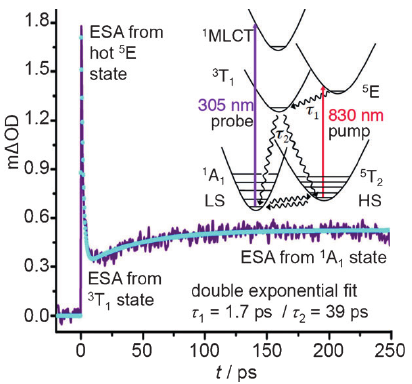
Light-induced excited spin state trapping (LIESST) in iron(II) spin-crossover compounds, i.e., the light-induced population of the high-spin (S=2) state below the thermal transition temperature, was discovered thirty years ago. For irradiation into metal-ligand charge transfer (MLCT) bands of the low-spin (S=0) species the acknowledged sequence takes the system from the initially excited 1MLCT to the high-spin state via the 3MLCT state within ~150 fs, thereby bypassing low-lying ligand-field (LF) states. Nevertheless, these play role, as borne out by the observation of LIESST and reverse-LIESST on irradiation directly into the LF bands for systems with only high-energy MLCT states. Herein we elucidate the ultrafast reverse-LIESST pathway by identifying the lowest energy S=1 LF state as intermediate state with a lifetime of 39 ps for the light-induced high-spin to low-spin conversion on irradiation into the spin-allowed LF transition of the high-spin species in the NIR. | ||||||||

 |
|
|||||||
For numerous spin crossover complexes, the anisotropic distortion of the first coordination shell around the transition metal center governs the dynamics of the high-spin/lowspin interconversion. However, this structural parameter remains elusive for samples that cannot be investigated with crystallography. The present work demonstrates how picosecond X-ray absorption spectroscopy is able to capture this specifi c deformation in the photoinduced high-spin state of solvated [Fe(terpy)2 ]2+ , a complex which belongs to the prominent family of spin crossover building blocks with nonequivalent metal− ligand bonds. The correlated changes in Fe−NAxial , Fe− NDistal , and bite angle NDistal− Fe− NAxial extracted from the measurements are in very good agreement with those predicted by DFT calculations in D2d symmetry. The outlined methodology is generally applicable to the characterization of ultrafast nuclear rearrangements around metal centers in photoactive molecular complexes and nanomaterials, including those that do not display long-range order. | ||||||||
|
 |
|||||||
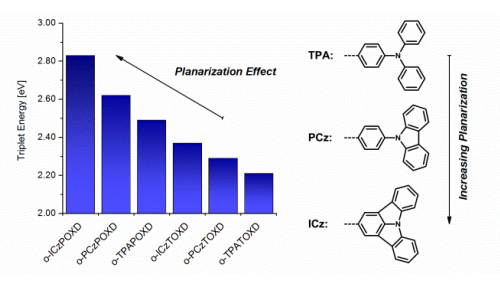
A series of 6 novel triarylamine-containing oxadiazole compounds (o-PCzPOXD, o-ICzPOXD, o-TPATOXD, o-PCzTOXD, o-ICzTOXD, o-CzTOXD) have been designed, synthesized and characterized concerning applications as host materials in PHOLED devices. To further improve the ortho-linkage concept, the impact of incorporating planarized electron-donating triarylamine (TAA) structures on intramolecular charge transfer was examined. The effect was evaluated for two series of electron-accepting oxadiazole scaffolds, realizing ortho-linkage on the benzene (POXD) and the thiophene (TOXD) core. Thermal analysis shows increased glass-transition temperatures for planarized structures indicating an improved morphological stability. A higher degree of planarization also results in significantly increased singlet and triplet energy values, revealing the impact on the intramolecular charge transfer. Employing the developed materials, red (o-TPATOXD: CEmax: 28.8 cd A-1, EQEmax: 16.9%), green (o-PCzPOXD: CEmax: 62.9 cd A-1, EQEmax: 17.1%) and blue (o-PCzPOXD: CEmax: 29.8 cd A-1, EQEmax: 13.4%) devices were achieved showing remarkably low efficiency roll-off for planarized donors. Hence, this is the first report of efficient blue devices for this specific class of host materials. It is proposed that the results correlate with an increasing ortho-linkage effect and decreasing donor strength of the TAA moiety by planarization and, thus, tackling one of the major challenges in PHOLED research: improving both triplet energy and compound stability. | ||||||||
|
||||||||
A large pi-conjugated chromophore composed of two dipyrido[3,2-a:2’,3’-c]phenazine (dppz) units directly fused to the central tetrathiafulvalene (TTF) core, has been prepared as a bridging ligand, and its strong binding ability to Ru2+ forming a new dinuclear complex is presented. The electronic absorption and luminescence and the electrochemical behaviour of the free ligand as well as the Ru2+ complex have been investigated in detail. The free ligand shows a very strong band in the UV region consistent with ligand centred π-π* transitions and an intense broad band in the visible region corresponding to an intramolecular charge transfer (ILCT) transition. Upon coordination, a metal-to-ligand charge transfer (MLCT) appears at 22520 cm-1 while the ILCT band is bathochromically shifted by 1620 cm-1. These electrochemically amphoteric chromophores have also been characterized by spectroelectrochemical methods. The oxidized radical species of the free ligand show a strong tendency to undergo aggregation, in which long-distance attractive interactions overcome the electrostatic repulsion. Moreover, these two new chromophores reveal an ILCT fluorescence with large solvent-dependent Stokes shifts and quantum efficiencies of 0.052 for the free ligand and 0.016 for its dinuclear Ru2+ complex in CH2Cl2. | ||||||||
|
||||||||
Using the study of the low-spin complex [Fe(bpy)3]2+ in the gas phase and in condensed phases as a guideline, we examine different aspects of the application of DFT to the study of transition metal complexes in the framework of spin crossover or related phenomena. | ||||||||
|
||||||||
The crystal structure of the third polymorph of dibenzylsquaramide (Portell, A. et al., 2009), (fig. 1) has been determined from laboratory X-ray powder diffraction data by means of direct space methods using the computing program FOX. (Favre-Nicolin and Černý, 2002) The structure resolution has not been straightforward due to several difficulties on the indexing process and in the space group assignment. The asymmetric unit contains two different conformers, which has implied an additional difficulty during the Rietveld (Rietveld, 1969) refinement. All these issues together with particular structural features of disquaramides are discussed. | ||||||||
 |
|
|||||||
Light-upconversion via stepwise energy transfer from a sensitizer to an activator exploits linear optics for converting low-energy infrared or near-infrared incident photons to higher energy emission occurring in the part of the electromagnetic spectrum ranging from visible to ultraviolet. Stepwise excitation is restricted to activators possessing intermediate long-lived excited states such as those found for trivalent lanthanide cations dispersed in solid-state matrices. When the activator is embedded in a molecular complex, efficient non-radiative relaxation processes usually reduce excited state lifetimes to such an extent that upconversion becomes too inefficient to be detected under practical excitation intensities. Theoretical considerations suggest that the combination of millisecond timescale sensitizers with a central lanthanide activator located in supramolecular complexes circumvents this bottleneck by creating a novel pathway reminiscent of the energy transfer upconversion mechanism observed in doped solids. Application of this novel concept to chromium/erbium pairs in discrete triple-stranded helicates demonstrates that strong-field trivalent chromium chromophores irradiated with near-infrared photons produce upconverted green erbium-centered emission both in the solid state and in solution. | ||||||||
|
 |
|||||||
The reaction of 4,5-bis(2'-cyano-ethylsulfanyl)-4',5'-dipropylthiotetrathiafulvalene with [Pt(phen)Cl2] (phen = 1,10-phenanthroline) with CsOH as base in CH3OH–THF affords the target complex 1 in 44% yield. This complex crystallizes in the monoclinic space group P21/c, M = 790.01, a = 12.1732(12), b = 15.851(2), c = 14.5371(16) Å, b = 107.693(12)˚, V = 2672.4(5) Å3 and Z = 4. It undergoes two reversible single-electron oxidation and two irreversible reduction processes. An intense electronic absorption band at 15200 cm-1 (658 nm) in CH2Cl2 is assigned to the intramolecular mixed metal/ligand-to-ligand charge transfer (LLCT) from a tetrathiafulvalene-extended dithiolate-based HOMO to a phenanthroline-based LUMO. This band shifts hypsochromically with increasing solvent polarity. Systematic changes in the optical spectra upon oxidation allow precise tuning of the oxidation states of 1 and reversible control over its optical properties. Irradiation of 1 at 15625 cm-1 (640 nm) in glassy solution below 150 K results in emission from the 3LLCT excited state. | ||||||||
 |
|
|||||||
Ultrafast transient absorption spectroscopy serves to identify the 3dd state as intermediate quencher state of the 3MLCT luminescence in the non-luminescent ruthenium complexes [Ru(m-bpy)3]2+ (m-bpy = 6-methyl-2,2′-bipyridine) and [Ru(tm-bpy)3]2+ (tm-bpy = 4,4′,6,6′-tetramethyl-2′,2′-bipyridine). For [Ru(m-bpy)3]2+, the population of the 3dd state from the 3MLCT state occurs within 1.6 ps, while the return to the ground state takes 450 ps. For [Ru(tm-bpy)3]2+, the corresponding values are 0.16 and 7.5 ps, respectively. According to DFT calculations, methyl groups added in the 6 and 6′ positions of bipyridine stabilize the 3dd state by ∼4000 cm–1 each, compared to [Ru(bpy)3]2+. | ||||||||
|
|
 |
|||||||
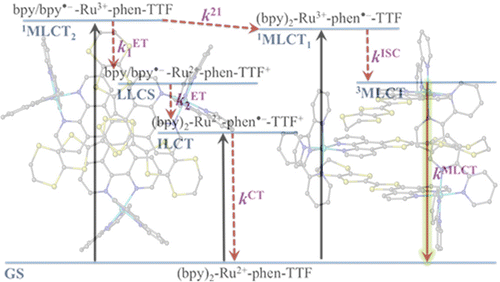
Ru(II) complexes with chelating ligands, 4′,5′-ethylenedithiotetrathiafulvenyl[4,5-f][1,10]phenanthroline (L1), 1,3-dithiole-2-thiono[4,5-f][1,10]phenanthroline (L2), and 1,3-dithiole-2-ono[4,5-f][1,10]phenanthroline (L3), have been prepared and their structural, electrochemical, and photophysical properties investigated. Density functional theory (DFT) calculations indicate that the highest occupied molecular orbital of [Ru(bpy)2(L1)](PF6)2 (1) is located on the tetrathiafulvalene (TTF) subunit and appears ≈0.6 eV above the three Ru-centered d orbitals. In agreement with this finding, 1 exhibits three reversible oxidations: the two at lower potentials take place on the TTF subunit, and the one at higher potential is due to the Ru3+/Ru2+ redox couple. Complexes [Ru(bpy)2(L2)](PF6)2 (2) and [Ru(bpy)2(L3)](PF6)2 (3) exhibit only the Ru3+/Ru2+-related oxidation. The optical absorption spectra of all complexes reveal a characteristic metal-to-ligand charge transfer (MLCT) band centered around 450 nm. In addition, in the spectrum of 1 the MLCT band is augmented by a low-energy tail that extends beyond 500 nm and is attributed to the intraligand charge transfer (ILCT) transition of L1, according to time-dependent DFT calculations. The substantial decrease in the luminescence quantum yield of 1 compared to those of 2 and 3 is attributed to the reductive quenching of the emissive state via electron transfer from the TTF subunit to the Ru3+ center, thus allowing nonradiative relaxation to the ground state through the lower-lying ILCT state. In the presence of O2, complex 1 undergoes a photoinduced oxidative cleavage of the central C═C bond of the TTF fragment, resulting in complete transformation to 3. This photodegradation process was studied with 13C NMR and optical absorption spectroscopy. | ||||||||
 |
|
|||||||
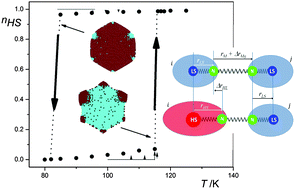
Whereas the neat polymeric Fe(II) compound {[Fe(bbtr)3](ClO4)2}∞ (bbtr=1,4-di(1,2,3-triazol-1-yl)butane) shows an abrupt spin transition centered at 107 K facilitated by a crystallographic symmetry breaking, in the covalently linked 2D coordination network of {[Fe(bbtr)3](BF4)2}∞, Fe(II) stays in the high-spin state down to 10 K. However, strong cooperative effects of elastic origin result in reversible, persistent and wavelength-selective photoswitching between the low-spin and high-spin manifolds. This compound thus shows true light-induced bistability below 100 K. The persistent bidirectional optical switching behavior is discussed as a function of temperature, irradiation time and intensity. Crystallographic studies reveal a photo-induced symmetry breaking and serve to establish the correlation between structure and cooperative effects. The static and kinetic behavior is explicated within the framework of the mean-field approximation. | ||||||||
|
||||||||
In the spin-crossover compound [Fe(6-mepy)3tren](PF6)2, (6-mepy)3tren = tris{4-[(6-methyl)-2-pyridyl]-3-aza-butenyl}amine, the high-spin state can be populated as metastable state below the thermal transition temperature via irradiation into the metal to ligand charge transfer absorption band of the low-spin species. At 10 K, the lifetime of this metastable state is only 1 s. Despite this, it is possible to determine an accurate excited state structure by following the evolution of relevant structural parameters by synchrotron X-ray diffraction under continuous irradiation with increasing intensity. The difference in metal-ligand bond length between the high-spin and the low-spin state is found to be 0.192 Å obtained from an analysis of the experimental data using the mean-field approximation to model cooperative effects. | ||||||||
|
||||||||
A new cyclen derivative L, bearing a methyl-chromeno-pyridinylidene hydrazone moiety, was synthesized and studied in MeOH, as potential fluorescent “OFF-on-ON” sensors for Zn(II). Photocphysical properties of this ligand being PET regulated, L was only weakly emissive in the absence of metal ions (OFF). L fluorescence was increased modestly upon addition of one equivalent of Zn(II), and further increased upon addition of a second equivalent. Therefore, Zn:L behaved as a highly sensitive ON sensor for zinc. This efficiency was correlated to Zn(II) coordination via the hydrazone moiety of the fluorophore, producing an efficient CHelation-Enhanced Fluorescence (CHEF) effect. A complementary theoretical study carried out with DFT calculations further elucidated of the optical properties. | ||||||||
|
||||||||
A switch in time: A fast precipitation technique was used to prepare 75 nm FeII spin-crossover nanocrystals. Their photoswitching dynamics, based on the light-induced excited spin-state trapping effect, has been investigated by means of optical spectroscopy. A significant variation of the switching proprieties is observed compared to similar but amorphous nanoparticles. | ||||||||
|
 |
|||||||
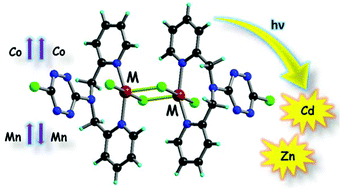
The insertion of a [Fe(sal2-trien)]+ complex cation into a 2D oxalate network in the presence of different solvents results in a family of hybrid magnets with coexistence of magnetic ordering and photoinduced spin-crossover (LIESST effect) in compounds [FeIII(sal2-trien)][MnIICrIII(ox)3]·CHCl3 (1·CHCl3), [FeIII(sal2-trien)][MnIICrIII(ox)3]·CHBr3 (1·CHBr3), and [FeIII(sal2-trien)][MnIICrIII(ox)3]·CH2Br2 (1·CH2Br2). The three compounds crystallize in a 2D honeycomb anionic layer formed by MnII and CrIII ions linked through oxalate ligands and a layer of [Fe(sal2-trien)]+ complexes and solvent molecules (CHCl3, CHBr3, or CH2Br2) intercalated between the 2D oxalate network. The magnetic properties and Mössbauer spectroscopy indicate that they undergo long-range ferromagnetic ordering at 5.6 K and a spin crossover of the intercalated [Fe(sal2-trien)]+ complexes at different temperatures T1/2. The three compounds present a LIESST effect with a relaxation temperature TLIESST inversely proportional to T1/2. The isostructural paramagnetic compound, [FeIII(sal2-trien)][ZnIICrIII(ox)3]·CH2Cl2 (2·CH2Cl2) was also prepared. This compound presents a partial spin crossover of the inserted FeIII complex as well as a LIESST effect. Finally, spectroscopic characterization of the FeIII doped compound [Ga0.99Fe0.01(sal2trien)][MnIICrIII(ox)3]·CH2Cl2 (3·CH2Cl2) shows a gradual and complete thermal spin crossover and a LIESST effect on the isolated FeIII complexes. This result confirms that cooperativity is not a necessary condition to observe the LIESST effect in an FeIII compound. | ||||||||
 |
|
|||||||
Four-dimensional (4D) electron microscopy (EM) uniquely combines the high spatial resolution to pinpoint individual nano-objects, with the high temporal resolution necessary to address the dynamics of their laser-induced transformation. Here, using 4D-EM, we demonstrate the in situ irreversible transformation of individual nanoparticles of the molecular framework Fe(pyrazine)Pt(CN)4. The newly formed material exhibits an unusually large negative thermal expansion (i.e. contraction), which is revealed by time-resolved imaging and diffraction. Negative thermal expansion is a unique property exhibited by only few materials. Here we show that the increased flexibility of the metal–cyanide framework after the removal of the bridging pyrazine ligands is responsible for the negative thermal expansion behavior of the new material. This in situ visualization of single nanostructures during reactions should be extendable to other classes of reactive systems. | ||||||||
 |
||||||||
The advancement of techniques that can probe the behaviour of individual nanoscopic objects is of paramount importance in various disciplines, including photonics and electronics. As it provides images with a spatiotemporal resolution, four-dimensional electron microscopy, in principle, should enable the visualization of single-nanoparticle structural dynamics in real and reciprocal space. Here, we demonstrate the selectivity and sensitivity of the technique by visualizing the spin crossover dynamics of single, isolated metal–organic framework nanocrystals. By introducing a small aperture in the microscope, it was possible to follow the phase transition and the associated structural dynamics within a single particle. Its behaviour was observed to be distinct from that imaged by averaging over ensembles of heterogeneous nanoparticles. The approach reported here has potential applications in other nanosystems and those that undergo (bio)chemical transformations. | ||||||||
|
 |
|||||||
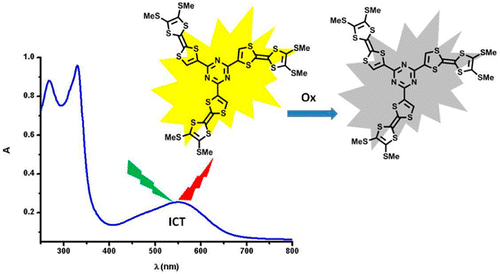
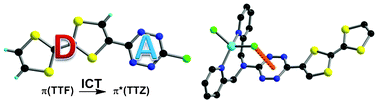
Palladium-catalyzed cross-coupling reactions between chlorinated 1,3,5-triazines (TZ) and tetrathiafulvalene (TTF) trimethyltin derivatives afford mono- and C3 symmetric tris(TTF)-triazines as donor–acceptor compounds in which the intramolecular charge transfer (ICT) is modulated by the substitution scheme on TTF and TZ and by chemical or electrochemical oxidation. The TTF-TZ-Cl2 and (SMe)2TTF-TZ-Cl2 derivatives show fully planar structures in the solid state as a consequence of the conjugation between the two units. Electrochemical and photophysical investigations, supported by theoretical calculations, clearly demonstrate that the lowest excited state can be ascribed to the intramolecular charge transfer (ICT) π(TTF)→π*(TZ) transition. The tris(TTF) compound [(SMe)2TTF]3-TZ shows fluorescence when excited in the ICT band, and the emission is quenched upon oxidation. The radical cations TTF+• are easily observed in all of the cases through chemical and electrochemical oxidation by steady-state absorption experiments. In the case of [(SMe)2TTF]3-TZ, a low energy band at 5000 cm–1, corresponding to a coupling between TTF+• and TTF units, is observed. A crystalline radical cation salt with the TTF-TZ-Cl2 donor and PF6– anion, prepared by electrocrystallization, is described. | ||||||||
 |
|
|||||||
A comparison of the vibrational spectra of many inorganic borohydrides allows us to distinguish compounds with isolated BH4- ions and compounds containing complex ions such as Sc(BH4)4-. The characteristic spectral features of both types of compounds are identified, showing that the B–H bonding is quite different in both cases. A detailed analysis of the vibrations of the isolated BH4- ions provides new information about their local structure. Angular deformations of individual borohydride ion are analyzed quantitatively. It appears that the compounds containing isolated BH4- ions belong to those with the most electropositive cations and the highest decomposition temperature, while the complex borohydrides show significantly lower decomposition temperatures and possible diborane formation. | ||||||||
|
||||||||
Two benzodifuran (BDF)-coupled spiropyran (SP) systems and their BDF reference compounds were obtained in good yields through Huisgen–Meldal–Sharpless “click” chemistry and then subjected to investigation of their electrochemical and photophysical properties. In both SP and merocyanine (MC) forms of the coupled molecules, the BDF-based emission is quenched to around 1 % of the quantum yield of emission from the BDF reference compounds. Based on electrochemical data, this quenching is attributed to oxidative electron-transfer quenching. Irradiation at 366 nm results in ring opening to the MC forms of the BDF-coupled SP compounds and the SP reference compound with a quantum efficiency of about 50 %. The rate constants for the thermal ring closing are approximately 3.4×10−3 s−1. However, in the photostationary states the MC fractions of the coupled molecules are substantially lower than that of the reference SP compound, attributed to the observed acceleration of the ring-closing reaction upon irradiation. As irradiation at 366 nm invariably also excites higher-energy transitions of the BDF units in the coupled compounds, the ring-opening reaction is accelerated relative to the SP reference, which results in lower MC fractions in the photostationary state. Reversible photochromism of these BDF-coupled SP compounds renders them promising in the field of molecular switches. | ||||||||
|
 |
|||||||
The structurally characterized tetrathiafulvalene-1,2,4,5-tetrazine donor–acceptor system shows redox tuneable intramolecular charge transfer, solvatochromic and electrochromic behaviour. Attachment of a dipicolyl-amine chelating unit affords a multifunctional ligand, which allows the preparation of the ZnCl2 complex in which an anion-π interaction is seen. | ||||||||
|
||||||||
Two pyridylphenols with intramolecular hydrogen bonds between the phenol and pyridine units have been synthesized, characterized crystallographically, and investigated by cyclic voltammetry and UV/Vis spectroscopy. Reductive quenching of the triplet metal-to-ligand charge-transfer excited state of the [Re(CO)3(phen)(py)]+ complex (phen=1,10-phenanthroline, py=pyridine) by the two pyridylphenols and two reference phenol molecules is investigated by steady-state and time-resolved luminescence spectroscopy, as well as by transient absorption spectroscopy. Stern–Volmer analysis of the luminescence quenching data provides rate constants for the bimolecular excited-state quenching reactions. H/D kinetic isotope effects for the pyridylphenols are on the order of 2.0, and the bimolecular quenching reactions are up to 100 times faster with the pyridylphenols than with the reference phenols. This observation is attributed to the markedly less positive oxidation potentials of the pyridylphenols with respect to the reference phenols (≈0.5 V), which in turn is caused by proton coupling of the phenol oxidation process. Transient absorption spectroscopy provides unambiguous evidence for the photogeneration of phenoxyl radicals, that is, the overall photoreaction is clearly a proton-coupled electron-transfer process. | ||||||||
 |
|
|||||||
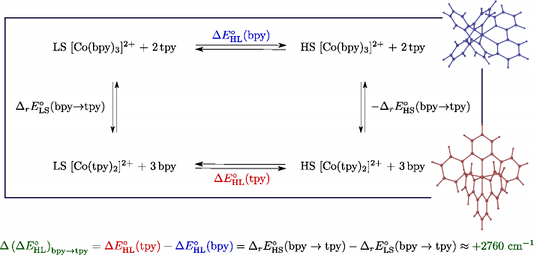
We report a detailed DFT study of the energetic and structural properties of the spin-crossover Co(II) complex [Co(tpy)2]2+ (tpy = 2,2′:6′,2′′-terpyridine) in the low-spin (LS) and the high-spin (HS) states, using several generalized gradient approximation and hybrid functionals. In either spin-state, the results obtained with the functionals are consistent with one another and in good agreement with available experimental data. Although the different functionals correctly predict the LS state as the electronic ground state of [Co(tpy)2]2+, they give estimates of the HS–LS zero-point energy difference ΔE0HL (tpy) which strongly depend on the functional used. This dependency on the functional was also reported for the DFT estimates of the zero-point energy difference ΔE0HL (bpy) in the HS complex [Co(bpy)3]2+ (bpy = 2,2′-bipyridine) [A. Vargas, A. Hauser and L. M. Lawson Daku, J. Chem. Theory Comput., 2009, 5, 97]. The comparison of the ΔE0HL (tpy) and ΔE0HL (bpy) estimates showed that all functionals correctly predict an increase of the zero-point energy difference upon the bpy → tpy ligand substitution, which furthermore weakly depends on the functionals, amounting to (ΔE0HL)bpy->tpy ≈ +2670 cm-1 . From these results and basic thermodynamic considerations, we establish that, despite their limitations, current DFT methods can be applied to the accurate determination of the spin-state energetics of complexes of a transition metal ion, or of these complexes in different environments, provided that the spin-state energetics is accurately known in one case. Thus, making use of the availability of a highly accurate ab initio estimate of the HS–LS energy difference in the complex [Co(NCH)6]2+ [L. M. Lawson Daku, F. Aquilante, T. W. Robinson and A. Hauser, J. Chem. Theory Comput., 2012, 8, 4216], we obtain for [Co(tpy)2]2+ and [Co(bpy)3]2+best estimates of ΔE0HL (bpy) ≈ -2800 cm-1 and ΔE0HL (tpy) ≈ 0 cm-1 , in good agreement with the known magnetic behaviour of the two complexes. | ||||||||
|
 |
|||||||
Electrochemical and photophysical analysis of new donor–acceptor systems 2 and 3, in which a benzothiadiazole (BTD) unit is covalently linked to a tetrathiafulvalene (TTF) core, have verified that the lowest excited state can be ascribed to an intramolecular-charge-transfer (ICT) π(TTF)→π*(benzothiadiazole) transition. Owing to better overlap of the HOMO and LUMO in the fused scaffold of compound 3, the intensity of the 1ICT band is substantially higher compared to that in compound 2. The corresponding CT fluorescence is also observed in both cases. The radical cation TTF+. is easily observed through chemical and electrochemical oxidation by performing steady-state absorption experiments. Interestingly, compound 2 is photo-oxidized under aerobic conditions. | ||||||||
 |
|
|||||||
The mechanoelastic model is applied to reproduce the experimental relaxation and thermal transition curves as determined for crystals of pure and diluted {[FexZn1–x(bbtr)3](ClO4)2}∞ [bbtr = 1,4-di(1,2,3-triazol-1-yl)butane] spin-crossover systems. In the mechanoelastic model, the spin-crossover complexes are situated in a hexagonal planar lattice, which is similar to the 2D coordination polymer with (3,6) network topology of [Fe(bbtr)3](ClO4)2. These complexes are linked by springs, which simulate the elastic interactions between them. Owing to the change in volume of the complexes during the spin transition, an elastic force accompanies the switch of every complex. This force propagates through the entire lattice and causes a shift of all molecules in the system and thus results in a new nuclear configuration. First, the ability of the model to reproduce various shapes of thermal transition and relaxation curves in pure compounds is analyzed; these range from gradual to very steep and include hysteresis behavior for the former and from single exponential to sigmoidal or with several steps for the latter. A structural phase transition can also be accounted for by changing the shape of the sample at a fixed temperature from a regular to an elongated hexagon. Furthermore, the effect of adding Zn as a dopant in a mixed crystal series is discussed. The role of dopants on the cluster evolution is also analyzed directly and by using the correlation factor. | ||||||||
|
 |
|||||||
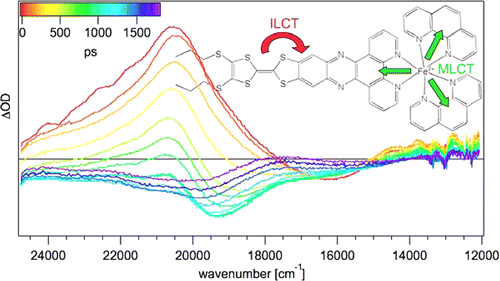
The synthesis and photophysical properties of the complex [Fe(phen)2(TTF-dppz)]2+ (TTF-dppz = 4′,5′-bis-(propylthio)tetrathiafulvenyl[i]dipyrido[3,2-a:2′,3′-c]phenazine, phen = 1,10-phenanthroline) are described. In this complex, excitation into the metal–ligand charge transfer bands results in the population of a high-spin state of iron(II), with a decay lifetime of approximately 1.5 ns, in dichloromethane, at room temperature. An intraligand charge transfer state can also be obtained and has a lifetime of 38 ps. A mechanism for the different states reached is proposed based on transient absorption spectroscopy. | ||||||||
 |
|
|||||||
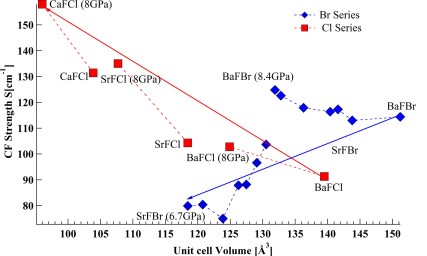
The emission spectra of Sm2+ doped in BaFBr and SrFBr hosts were measured at 10 K from ambient pressure to 8 GPa. The crystal field energy levels determined from the emission spectra were used to extract the free ion parameters (Fk and ζ ) and crystal field parameters (Bqk). The variation of Fk and ζ as a function of pressure was studied systematically and was discussed in relation to the central field and symmetry restricted covalency models. The change of the spin orbit coupling parameter (ζ) with pressure for SrFBr:Sm2+ showed very different behavior than in other matlockite hosts. Moreover the variation of Bqk under pressure was studied. The pressure dependence of the Bqk was described quantitatively using the Superposition Model (SM) with the help of structural parameters as a function of pressure, obtained from periodic DFT calculations. The validity of the SM was tested for Sm2+ in BaFBr and SrFBr. It is shown that this model does not apply to SrFBr, in contrast to other matlockite host materials. | ||||||||
|
 |
|||||||
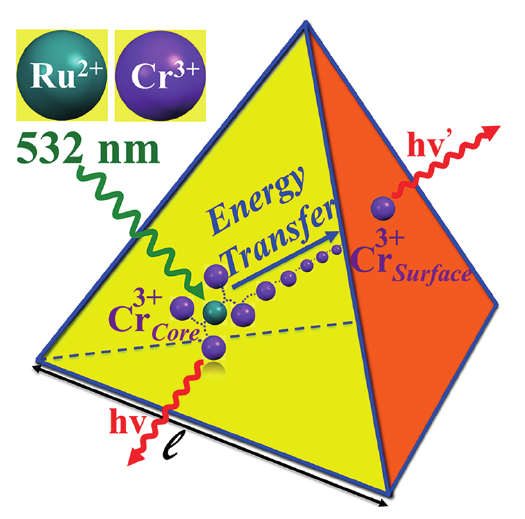
Chromium(III)-trisoxalate,[Cr(ox)3]3- (ox = C2O42-), incorporated into polymeric networks of composition [NaCr(ox)3][MII(bpy)3] and [NaCr(ox)3][MIII(bpy)3]ClO4 (bpy= 2,2'-bipyridine, MII = Zn, Fe, Ru; MIII = Rh, Cr), results in interesting features ranging from phonon-assisted and resonant energy migration within the R1 line the 2E state to persistent spectral side-hole burning via the latter, and manifestations of specific nearest-neighbour π–π interactions between bipyridine and oxalate. | ||||||||
 |
|
|||||||
Three iron(II) complexes, [Fe(TPMA)(BIM)](ClO4)2⋅0.5H2O (1), [Fe(TPMA)(XBIM)](ClO4)2 (2), and [Fe(TPMA)(XBBIM)](ClO4)2 ⋅0.75CH3OH (3), were prepared by reactions of FeII perchlorate and the corresponding ligands (TPMA=tris(2-pyridylmethyl)amine, BIM=2,2′-biimidazole, XBIM=1,1′-(α,α′-o-xylyl)-2,2′-biimidazole, XBBIM=1,1′-(α,α′-o-xylyl)-2,2′-bibenzimidazole). The compounds were investigated by a combination of X-ray crystallography, magnetic and photomagnetic measurements, and Mössbauer and optical absorption spectroscopy. Complex 1 exhibits a gradual spin crossover (SCO) with T1/2=190 K, whereas 2 exhibits an abrupt SCO with approximately 7 K thermal hysteresis (T1/2=196 K on cooling and 203 K on heating). Complex 3 is in the high-spin state in the 2–300 K range. The difference in the magnetic behavior was traced to differences between the inter- and intramolecular interactions in 1 and 2. The crystal packing of 2features a hierarchy of intermolecular interactions that result in increased cooperativity and abruptness of the spin transition. In 3, steric repulsion between H atoms of one of the pyridyl substituents of TPMA and one of the benzene rings of XBBIM results in a strong distortion of the FeII coordination environment, which stabilizes the high-spin state of the complex. Both 1 and 2 exhibit a photoinduced low-spin to high-spin transition (LIESST effect) at 5 K. The difference in the character of intermolecular interactions of 1 and 2 also manifests in the kinetics of the decay of the photoinduced high-spin state. For 1, the decay rate constant follows the single-exponential law, whereas for 2 it is a stretched exponential, reflecting the hierarchical nature of intermolecular contacts. The structural parameters of the photoinduced high-spin state at 50 K are similar to those determined for the high-spin state at 295 K. This study shows that N-alkylation of BIM has a negligible effect on the ligand field strength. Therefore, the combination of TPMA and BIM offers a promising ligand platform for the design of functionalized SCO complexes. | ||||||||
|
||||||||
Highly accurate estimates of the high-spin/low-spin energy difference ΔEHLel in the high-spin complexes [Fe(NCH)6]2+ and [Co(NCH)6]2+ have been obtained from the results of CCSD(T) calculations extrapolated to the complete basis set limit. These estimates are shown to be strongly influenced by scalar relativistic effects. They have been used to assess the performances of the CASPT2 method and of 30 density functionals of the GGA, meta-GGA, global hybrid, RSH and double-hybrid types. For the CASPT2 method, the results of the assessment support the proposal [Kepenekian, M.; Robert, V.; Le Guennic, B. J. Chem. Phys.2009, 131, 114702] that the ionization potential–electron affinity (IPEA) shift defining the zeroth-order Hamiltonian be raised from its standard value of 0.25 au to 0.50–0.70 au for the determination of ΔEHLel in Fe(II) complexes with a [FeN6] core. At the DFT level, some of the assessed functionals proved to perform within chemical accuracy (±350 cm-1) for the spin-state energetics of [Fe(NCH)6]2+, others for that of [Co(NCH)6]2+, but none of them simultaneously for both complexes. As demonstrated through a reparametrization of the CAM-PBE0 range-separated hybrid, which led to a functional that performs within chemical accuracy for the spin-state energetics of both complexes, performing density functionals of broad applicability may be devised by including in their training sets highly accurate data like those reported here for [Fe(NCH)6]2+ and [Co(NCH)6]2+. | ||||||||
|
 |
|||||||
The thermal spin transition, the photoexcitation, and the subsequent spin relaxation in the mixed crystal series of the covalently linked two-dimensional network {[Zn1-xFex(bbtr)3](ClO4)2}∞ (x = 0.02–1, bbtr =1,4-di(1,2,3-triazol-1-yl)-butane) are discussed. In the neat compound, the thermal spin transition with a hysteresis of 13 K is accompanied by a crystallographic phase transition (Kusz, J.; Bronisz, R.; Zubko, M.; Bednarek, H. Chem. Eur. J.2011, 17, 6807). In contrast, the diluted crystals with x ≤ 0.1 stay essentially in the high-spin state down to low temperatures and show typical first order relaxation kinetics upon photoexcitation, and the structural phase transition is well separated from the spin transition. With increasing Fe(II) concentration, steeper thermal transitions and sigmoidal relaxation curves indicate increasingly important cooperative effects. Already at x = 0.38, the spin relaxation is governed by cooperative interactions between Fe(II) centers, and the crystallographic phase transition begins to influence the spin transition. The kinetic behavior of the thermal spin transition is reproduced within the framework of a dynamic mean-field model. | ||||||||
|
||||||||
A new room-temperature chromium tricarbonyl-mediated cycloaromatization of enediynes is reported. The reaction occurs with both cyclic and acyclic enediynes in the presence of [Cr(CO)3(η6-naphthalene)] and both a coordinating solvent and a hydrogen atom source, providing chromium–arene complexes in reasonable yield and good diastereocontrol. The mechanism of the reaction has been probed through DFT computational and spectroscopic methods. These studies suggest that direct C1–C6 bond formation from an η6-enediyne complex is the lowest-energy path, forming a metal-bound p-benzyne biradical. NMR spectroscopy suggests that enediyne alkene coordination occurs in preference to alkyne coordination, forming a THF-stabilized olefin intermediate; subsequent alkyne coordination leads to cyclization. While biradical quenching occurs rapidly and primarily via the singlet biradical, the triplet state biradical is detectable by EPR spectroscopy, suggesting intersystem crossing to a triplet ground state. | ||||||||
 |
|
|||||||
This work illustrates a simple approach for optimizing long-lived near-infrared lanthanide-centered luminescence using trivalent chromium chromophores as sensitizers. Reactions of the segmental ligand L2 with stoichiometric amounts of M(CF3SO3)2 (M = Cr, Zn) and Ln(CF3SO3)3 (Ln = Nd, Er, Yb) under aerobic conditions quantitatively yield the D3-symmetrical trinuclear [MLnM(L2)3](CF3SO3)n complexes (M = Zn, n = 7; M = Cr, n = 9), in which the central lanthanide activator is sandwiched between the two transition metal cations. Visible or NIR irradiation of the peripheral Cr(III) chromophores in [CrLnCr(L2)3]9+ induces rate-limiting intramolecular intermetallic Cr→Ln energy transfer processes (Ln = Nd, Er, Yb), which eventually produces lanthanide-centered near-infrared (NIR) or IR emission with apparent lifetimes within the millisecond range. As compared to the parent dinuclear complexes [CrLn(L1)3]6+, the connection of a second strong-field [CrN6] sensitizer in [CrLnCr(L2)3]9+ significantly enhances the emission intensity without perturbing the kinetic regime. This work opens novel exciting photophysical perspectives via the buildup of non-negligible population densities for the long-lived doubly excited state [Cr*LnCr*(L2)3]9+ under reasonable pumping powers. | ||||||||
|
 |
|||||||
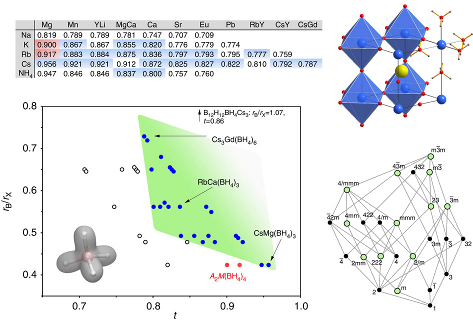
Four novel bimetallic borohydrides have been discovered, K2M(BH4)4 (M = Mg or Mn), K3Mg(BH4)5, and KMn(BH4)3, and are carefully investigated structurally as well as regarding their decomposition reaction mechanism by means of in situ synchrotron radiation powder X-ray diffraction (SR-PXD), vibrational spectroscopies (Raman and IR), thermal analysis (TGA and DTA), and ab initio density functional theory (DFT) calculations. Mechano-chemical synthesis (ball-milling) using the reactants KBH4, α-Mg(BH4)2, and α-Mn(BH4)2 ensures chlorine-free reaction products. A detailed structural analysis reveals significant similarities as well as surprising differences among the two isomorphs K2M(BH4)4, most importantly concerning the extent to which the complex anion [M(BH4)4]2– is isolated in the structure. Anisotropic thermal expansion and an increase in symmetry at high temperatures in K3Mg(BH4)5 is ascribed to the motion of BH4 groups inducing hydrogen repulsive effects, and the dynamics of K3Mg(BH4)5 are investigated. Decomposition in the manganese system proceeds via the formation of KMn(BH4)3, the first perovkite type borohydride reported to date. | ||||||||
 |
|
|||||||
Colloidal Mn2+-doped semiconductor nanocrystals such as Mn2+:ZnSe have attracted broad attention for potential applications in phosphor and imaging technologies. Here, we report saturation of the sensitized Mn2+ photoluminescence intensity at very low continuous-wave (CW) and quasi-CW photoexcitation powers under conditions that are relevant to many of the proposed applications. Time-resolved photoluminescence measurements and kinetic modeling indicate that this saturation arises from an Auger-type nonradiative cross relaxation between an excited Mn2+ ion and an exciton within the same nanocrystal. A lower limit of k = 2 × 1010 s–1 is established for the fundamental rate constant of the Mn2+(4T1)-exciton cross relaxation. | ||||||||
|
||||||||
This contribution investigates LnIII complexes formed with a small ditopic ligand, L1, and their structural, thermodynamic and photophysical properties. The spectrophotometric and NMR titrations evidence the triangular assemblies [Ln3(L1-H)3]6+ at stoichiometric conditions and their properties are discussed in relation to L2-containing analogues. In addition, the dinuclear species, [Ln2(L1-H)]5+, is observed with an excess of metal. | ||||||||
|
||||||||
Due to its extreme kinetic inertness, trivalent chromium, Cr(III), has been rarely combined with labile trivalent lanthanides, Ln(III), to give discrete self-assembled (supra)molecular polynuclear complexes. However, the plethora of accessible metal-centered excited states possessing variable lifetimes and emissive properties, combined with the design of efficient intramolecular Cr(III) ↔ Ln(III) energy transfer processes open attractive perspectives for programming directional light-conversion within these heterometallic molecules. Efforts made to address this exciting challenge for both light-sensitization and light-upconversion are discussed in this article. | ||||||||
|
 |
|||||||
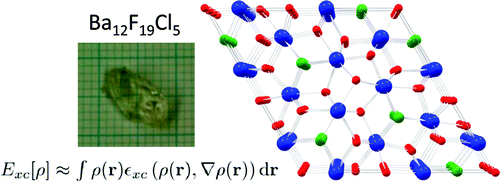
The crystal chemistry of the barium fluoride chloride system is studied both experimentally and theoretically. Different synthetic approaches yield nanocrystalline materials as well as large single crystals. The crystalline phases identified so far are BaFCl, Ba12F19Cl5 and Ba7F12Cl2 (in two modifications) and compared with analogous compounds. It is demonstrated that the compound Ba2F3Cl reported by Fessenden and Lewin 50 years ago corresponds to Ba7F12Cl2. The phase diagram of the BaCl2 – BaF2 system is reinvestigated for fluoride mole fractions between 0.5 and 1. The peritectic formation of Ba12F19Cl5 is observed. Periodic DFT calculations are performed for all structures in this system, including a hypothetical structure for Ba2F3Cl, based on the experimental structure of Ba2H3Cl. The energy of formation of the different barium fluoride chloride compounds from BaCl2 and BaF2 (normalized for one barium atom per formula unit), as calculated by DFT at 0K, is within only about ± 15 kJ/mol. Comparison with recent experimental results on calcium and strontium hydride chloride (bromide) compounds, suggest the possibility of a mutual exclusion between the M2X3Y and M7X12Y2 (M = Ca, Sr, Ba, Pb, X = H, F, Y = Cl,Br) structures. The single crystal structure of PbFBr is also reported. | ||||||||
 |
|
|||||||
In the covalently linked 2D coordination network {[Fe(bbtr)3](BF4)2}∞, bbtr = 1,4-di(1,2,3-triazol-1-yl)butane, the iron(II) centers stay in the high-spin (HS) state down to 10 K. They can, however, be quantitatively converted to the low-spin (LS) state by irradiating into the near-IR spin allowed 5dd band and back again by irradiating into the visible 1dd band. The compound shows true light-induced bistability below 100 K, thus, having the potential for persistent bidirectional optical switching at elevated temperatures. | ||||||||
|
 |
|||||||
Dicyanoaurate reacts with the organic acceptor molecule, 1,1′-bis-(2,4-dinitrophenyl)-4,4′-bipyridinium, DNP, to form a supramolecular complex with the general formula {[Au(CN)2]2DNP}·4H2O. The complex was characterized by X-ray crystallography, and its photophysical properties were investigated in the solid-state. Although the initial (DNP)Cl2 compound does not show photoluminescence behavior and the dicyanoaurate shows photoluminescence only in the UV range, the resulting supramolecular complex displays two simultaneous, essentially independent, photoluminescence bands in the visible range originating from individual contributions of the DNP unit and the dicyanoaurate dimers. This unusual simultaneous photoluminescence behavior displayed by both the dicyanoaurate donor units and the redox-active 4,4′-bipyridinium acceptor have lifetimes of 0.5 μs and several hundred μs, respectively. | ||||||||
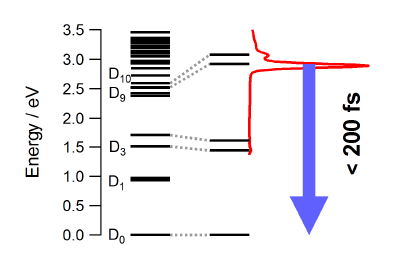 |
|
|||||||
The photophysical properties of the free neutral radical galvinoxyl were studied by a combination of femtosecond time-resolved spectroscopy and quantum chemical calculations. The electronic absorption spectrum is dominated by an intense band at 430 nm that is ascribed to the D9,10←D0 transitions. Upon photoexcitation at 400 nm, the population of the D9,10 states decays within less than 200 fs to the electronic ground state. This ultrafast internal conversion does not involve intramolecular modes with large amplitude motion as the measured dynamics does not show any significant dependence on the environment, but is most probably facilitated by a high density of electronic states of different character. Depending on the solvent, a weak transient band due to the galvinoxylate anion is also observed. This closed-shell species, which is fluorescent although its deactivation is also dominated by non-radiative decay, is generated upon biphotonic ionization of the solvent and electron capture. The ultrashort excited-state lifetime of the galvinoxyl radical precludes photoinduced disproportionation previously claimed to be at the origin of the formation of both anion and cation. | ||||||||
|
 |
|||||||
The recently developed mechanoelastic model is applied to characterize the thermal transition in spin-crossover complexes, with special attention given to the case of spin-crossover nanoparticles. In a two-dimensional system, hexagonal-shaped samples with open boundary conditions are composed of individual molecules that are linked by springs and can switch between two states, namely, the high-spin (HS) and the low-spin (LS) states. The switching of an individual molecule during the spin transition is decided by way of a Monte Carlo standard procedure, using transition probabilities depending on the temperature, the energy gap between the two states, the enthalpy difference, the degeneracy ratio, and the local pressure determined by the elongation or compression of its closest springs. The influence of external parameters, such as temperature sweeping rate and pressure, or intrinsic features of the system, such as the value of its spring constant, on the width of the thermal hysteresis, its shape, and its position are discussed. The particular case of spin-crossover nanoparticles is treated by considering them embedded into a polymer environment, which essentially affects the molecules situated at the edges and faces by decreasing their transition probabilities from HS to LS. Finally, the pressure hysteresis, obtained by varying the external pressure at constant temperature is discussed. | ||||||||
 |
|
|||||||
An efficient synthetic approach to a symmetrically functionalized tetrathiafulvalene (TTF) derivative with two diamine moieties, 2-[5,6-diamino-4,7-bis(4-pentylphenoxy)-1,3-benzodithiol-2-ylidene]-4,7-bis(4-pentylphenoxy)-1,3-benzodithiole-5,6-diamine (2), is reported. The subsequent Schiff-base reactions of 2 afford large π-conjugated multiple D–A arrays, for example the triad 2-[4,9-bis(4-pentylphenoxy)-1,3-dithiolo[4,5-g]quinoxalin-2-ylidene]-4,9-bis(4-pentylphenoxy)-1,3-dithiolo[4,5-g]quinoxaline (8) and the corresponding tetrabenz[bc,ef,hi,uv]ovalene-fused pentad 1, in good yields and high purity. The novel redox-active nanographene 1 is so far the largest known TTF-functionalized polycyclic aromatic hydrocarbon with a well-resolved 1H NMR spectrum. The electrochemically highly amphoteric pentad 1 and triad 8 exhibit various electronically excited charge-transfer states in different oxidation states leading to intense optical intramolecular charge transfer (ICT) absorbances over a wide spectral range. The chemical and electrochemical oxidations of 1 result in an unprecedented TTF•+ radical cation dimerization, leading to the formation of [1•+]2 at room temperature in solution due to the stabilizing effect arising from strong π–π interactions. Moreover, ICT fluorescence is observed with large solvent-dependent Stokes shifts and quantum efficiencies of 0.05 for 1 and 0.035 for 8 in CH2Cl2. | ||||||||
|
||||||||
We model here the behavior of spin transition compounds, considering molecules arranged in a 2D hexagonal lattice and interacting via springs. The role of impurities in the clustering and nucleation phenomena is analyzed, as well as the manner in which the impurities affect the relaxation curves. The switching of the individual molecules is checked using a Monte Carlo procedure. When a molecule changes its state, it also modifies its volume, and the new equilibrium positions of all the molecules are calculated. As in previously reported experiments, it is found here that bigger impurities slow down the relaxation from the metastable high-spin state to the low-spin state, while smaller impurities act in an opposite way. It is shown that if the concentration of the impurities is higher than a certain threshold, then they act as a barrier, trammeling the fast evolution of domains developing from the edges. | ||||||||
|
 |
|||||||
In this article, the synthesis of a novel high-conjugated ligand and its corresponding Ru(II) complex PTFTF:Ru is reported, along with the linear and nonlinear optical characterizations. Two-photon absorption based optical power limiting properties (OPL), especially in the near infrared, are described and compared to those of the analogous complexes previously published. Combined with a preliminary theoretical approach, this allows us to highlight several key parameters for OPL optimization in such molecular systems and more particularly the spectral overlap between TPA and excited-state absorption. | ||||||||
|
||||||||
Self-assembly processes between a tripodal ligand and LnIII cations have been investigated by means of supramolecular analytical methods. At an equimolar ratio of components, tetranuclear tetrahedral complexes are readily formed in acetonitrile. The structural analysis of the crystallographic data shows a helical wrapping of binding strands around metallic cations. The properties of this series of highly charged 3D compounds were examined by using NMR spectroscopy and optical methods in solution and in the solid state. In the presence of excess metal, a new trinuclear complex was identified. The X-ray crystal structure elucidated the coordination of metallic cations with two ligands of different conformations. By varying the metal/ligand ratio, a global speciation of this supramolecular system has been evidenced with different spectroscopic methods. In addition, these rather complicated equilibria were successfully characterised with the thermodynamic stability constants. A rational analysis of the self-assembly processes was attempted by using the thermodynamic free energy model and the impact of the ligand structure on the effective concentration is discussed. | ||||||||
 |
|
|||||||
The structure and thermodynamic properties of lanthanide complexes with a new tripodal ligand L2 have been elucidated using different physicochemical methods. At stoichiometric ratios, the tetrahedral three-dimensional complexes with lanthanide cations are formed in acetonitrile with good stabilities. Despite minor structural changes comparing to previously investigated tripodal ligands, the resulting assembly exhibits different features revealed with the crystal structure of [Eu4L24](OH)(ClO4)11 (orthorhombic, Pbcn). Interestingly, the highly charged edifice contains an inner cage encapsulating a perchlorate anion. Such lanthanide mediated cage-like assemblies are rare, and may be of interest for different sensing applications. Indeed, the anionic guest can be exchanged with different anions. The related host–guest equilibria were investigated with NMR techniques. Various aspects of these reactions are qualitatively discussed. | ||||||||
|
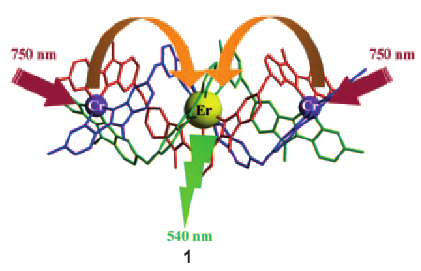 |
|||||||
The connection of two CrIII sensitizers around a central ErIII acceptor in a self-assembled cation provides high local metal concentrations that favor efficient nonlinear energy transfer upconversion luminescence (see picture). Upon selective low-energy near-infrared irradiation of CrIII-centered transitions, 1 displays an unprecedented molecular two-photon upconverted green ErIII-centered emission. | ||||||||
|
||||||||
We investigated by optical microscopy the thermal spin transition in single crystals of [Fe(bbtr)3](ClO4)2 (bbtr = 1,4-di(1,2,3-triazol-1-yl) butane). The growth of the low-spin phase was observed for different crystal orientations and sizes. The process always started from a corner of the crystal but its further development depended on the size, shape and thermal history of the crystal. In crystals of smaller size, under isothermal conditions, the low-spin phase developed in a continuous way, through the propagation of a rather well defined transformation front, with a higher propagation velocity inside the planes perpendicular to the c axis. In larger crystals the spontaneous occurrence of inhomogeneous stresses led to a stepwise propagation process. | ||||||||
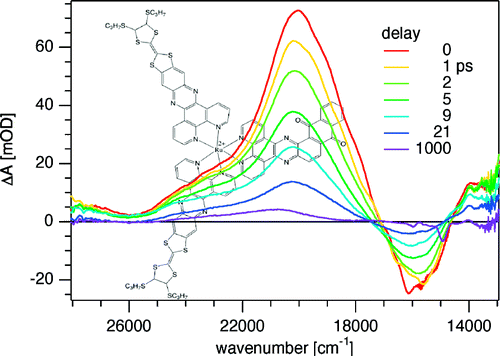 |
|
|||||||
The synthesis and the photophysical properties of the complex [Ru(TTF-dppz)2(Aqphen)]2+(TTF = tetrathiafulvalene, dppz = dipyrido-[3,2-a:2′,3′-c]phenazine, Aqphen = anthraquinone fused to phenanthroline via a pyrazine bridge) are described. In this molecular triad excitation into the metal−ligand charge transfer bands results in the creation of a long-lived charge separated state with TTF acting as electron donor and anthraquinone as terminal acceptor. The lifetime of the charge-separated state is 400 ns in dichloromethane at room temperature. A mechanism for the charge separation involving an intermediate charge-separated state is proposed based on transient absorption spectroscopy. | ||||||||
|
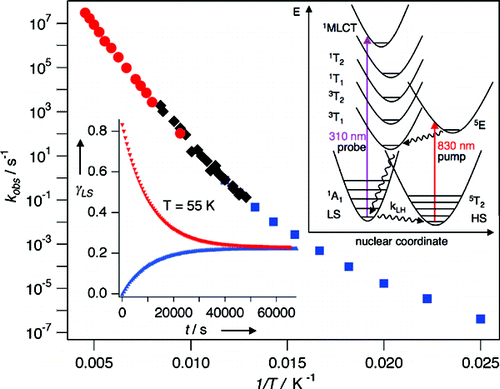 |
|||||||
Whereas the neat polymeric iron(II) compound [Fe(bbtr)3](ClO4)2, bbtr = 1,4-di(1,2,3-triazol-1-yl)butane, shows a quantitative spin transition triggered by a crystallographic phase transition centered at 107 K with a 13 K wide hysteresis, the iron(II) complexes in the diluted mixed crystals [FexZn1−x(bbtr)3](ClO4)2, x = 0.02 and 0.1, stay predominantly in the 5T2 high-spin state down to cryogenic temperatures. However, the 1A1 low-spin state can be populated as metastable state via irradiation into the spin-allowed 5T2→5E ligand-field transition of the high-spin species in the near-infrared. The quantum efficiency of the light-induced conversion is approximately 10% at low temperatures and decreases rapidly above 160 K. The lifetime of the light-induced low-spin state decreases from 15 days at 40 K to 30 ns at 220 K, that is, by 14 orders of magnitude. In the high-temperature regime the activation energy for the low-spin→high-spin relaxation is 1840(20) cm−1. | ||||||||
|
||||||||
The structural and vibrational properties of the isostructural compounds Ca2FeH6 and Sr2RuH6 are determined by periodic DFT calculations and compared with their previously published experimental crystal structures as well as new experimental vibrational data. The analysis of the vibrational data is extended to the whole series of alkaline-earth iron and ruthenium hydrides A2TH6 (A = Mg,Ca,Sr; T = Fe, Ru) in order to identify correlations between selected frequencies and the T-H bond length. The bulk moduli of Ca2FeH6 and Sr2RuH6 have also been determined within DFT. Their calculated values prove to compare well with the experimental values reported for Mg2FeH6 and several other compounds of this structure. | ||||||||
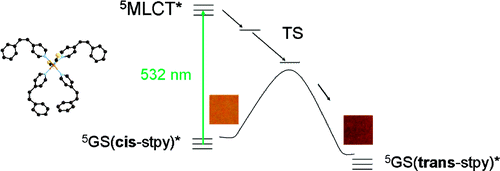 |
|
|||||||
The photoreactivity of two iron(II)−styrylpyridine frameworks Fe(stpy)4(NCSe)2 (stpy = 4-styrylpyridine) has been investigated for the very first time in a crystalline solid. A quantitative cis-to-trans isomerization of stilbenoids is shown to occur in the confined environment of the inorganic solid. The photochromic reaction was driven by a visible excitation into the metal-to-ligand charge transfer absorption of the high-spin all-cis complex. The solid-state transformation is accompanied by a unit-cell volume increase and an amorphization. Interestingly, the photoproduct formed by irradiating the high-spin all-cis reactant undergoes a spin conversion when the temperature is decreased. This observation is related to the “ligand-driven light-induced spin change” effect in a constrained environment. | ||||||||
|
 |
|||||||
Two tridentate and one bidentate binding strands have been anchored on a carbon atom to provide a new unsymmetrical tripodal ligand L for Ln(III) coordination. The ligand itself adopts a single conformation in solution stabilized by intramolecular hydrogen bonds evidenced in the solid state. The reaction of L with trivalent lanthanides provides different coordination complexes depending on the metal/ligand ratio. The speciation studies with selected lanthanides were performed in solution by means of NMR, ESMS, and spectrophotometric titrations. Differences in coordination properties along the lanthanide series were evidenced and may be associated with the changes in the ionic size. However, thermodynamic stability constants for the species of the same stoichiometry do not significantly vary. In addition, the structure of the dinuclear complex [Eu2L2]6+ has been elucidated in the solid state, where the complex crystallizes predominantly as an M-isomer. The crystal structure shows the coordination of two different ligands to each europium cation through tridentate strands, and the europium nine-coordinate sphere is completed with three solvent molecules. Finally, the results of photophysical investigations of [Eu2L2]6+ are in close agreement with the structural parameters determined by crystallography. | ||||||||
|
||||||||
In the dilute mixed-crystal system [Zn1−xFex(bbtr)3](ClO4)2, x=2 % (bbtr=1,4-di(1,2,3-triazol-1-yl)butane), the iron(II) centers are predominantly in the high-spin state. The low-spin state can be populated as a metastable state by irradiation with near-IR light; the rate constant of the low-spin→high-spin relaxation spans 14 orders of magnitude between 40 and 220 K | ||||||||
|
||||||||
In this paper we use a recently proposed elastic model in order to study the competition between linear photoexcitation and cooperative relaxation in spin-crossover molecular magnets. The difference in molecular size between the two possible spin states, that is, the high-spin and the low-spin states, respectively, induces distortions of the crystal lattice. These determine the elastic interactions between molecules, treated here as connecting springs that are either compressed or extended from their equilibrium length, thus modulating the local probability for the high-spin→low-spin relaxation. The crossover of individual molecules within the lattice is checked by a standard Monte Carlo procedure. Using very simple assumptions and a minimum number of parameters, photoexcitation curves and hysteresis loops under continuous irradiation below the thermal transition temperature can thus be simulated. The formation of clusters is analyzed and the presence of inhomogeneities in the system is investigated. | ||||||||
 |
|
|||||||
In this paper we study the cluster formation and evolution in spin crossover systems during the thermal transition in the frame of a mechano-elastic model applied to open boundary hexagonal lattices. The switching processes between the high-spin (HS) and low-spin (LS) state are studied by a method combining a Monte Carlo standard procedure on the spin state and the lattice relaxation. In the present study, we adopt the transition probabilities of the spin state taking into account the energy gap between the two states, the degeneracy ratio and the local pressure determined by the elongations of the closest springs. It is found that clusters of molecules in the same state tend to grow starting from corners, as in available experimental data. Some qualitative differences between the processes of cluster formation for the two hysteresis branches, i.e., HS to LS and LS to HS are pointed out. Moreover, we have studied the dependence of cluster formation on the strength of the elastic interactions, and also on the system size. The size dependence of the ratio between the system size and the maximum cluster length is very weak, which indicates the appearance of macroscopic domains. | ||||||||
|
||||||||
Resonant excitation energy transfer from [Cr(ox)3]3- to [Cr(bpy)3]3+ in the doped 3D oxalate networks [Rh1-xCrx(bpy)3][NaMIII1-yCry(ox)3]ClO4 (ox=C2O4-, bpy=2,2’-bipyridine, M=Al,Rh) is due to two types of interaction, namely super exchange coupling and electric dipole–dipole interaction. The energy transfer probability for both mechanisms is proportional to the spectral overlap of the 2E→4A2 emission of the [Cr(ox)3]3- donor and the 4A2→2T1 absorption of the [Cr(bpy)3]3+ acceptor.The spin-flip transitions of (pseudo-)octahedral Cr3+ are known to shift to lower energy with increasing pressure. Because the shift rates of the two transitions in question differ, the spectral overlap between the donor emission and the acceptor absorption is a function of applied pressure. For [Rh1-xCrx(bpy)3][Na-M1-yCry(ox)3]ClO4 the spectral overlap is thus substantially reduced on increasing pressure from 0 to 2.5 GPa. As a result, the energy transfer probability decreases with increasing pressure as evidenced by a decrease in the relative emission intensity from the [Cr(bpy)3]3+ acceptor. | ||||||||
|
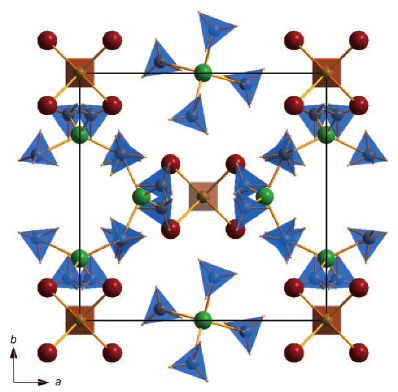 |
|||||||
The new double-cation Al-Li-borohydride is an attractive candidate material for hydrogen storage due to a very low hydrogen desorption temperature (~70 °C) combined with a high hydrogen density (17.2 wt %). It was synthesised by high-energy ball milling of AlCl3 and LiBH4. The structure of the compound was determined from image-plate synchrotron powder diffraction supported by DFT calculations. The material shows a unique 3D framework structure within the borohydrides (space group=P-43n, a=11.3640(3) Å). The unexpected composition Al3Li4(BH4)13 can be rationalized on the basis of a complex cation [(BH4)Li4]3+ and a complex anion [Al(BH4)4]-. The refinements from synchrotron powder diffraction of different samples revealed the presence of limited amounts of chloride ions replacing the borohydride on one site. In situ Raman spectroscopy, differential scanning calorimetry (DSC), thermogravimetry (TG) and thermal desorption measurements were used to study the decomposition pathway of the compound. Al-Li-borohydride decomposes at ~70 °C, forming LiBH4. The high mass loss of about 20 % during the decomposition indicates the release of not only hydrogen but also diborane. | ||||||||
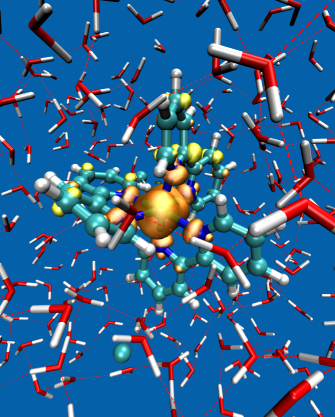 |
|
|||||||
The mechanism of the photoinduced low-spin → high-spin spin crossover is actively being investigated in Fe(II) complexes in solution using ultrafast spectroscopies. These studies accurately inform on the reaction coordinate of the Fe(II) chromophore upon photoexcitation. However, they leave open questions regarding the role of the solvent. Here, we report the description from a fully ab initio molecular dynamics study of the structure of [Fe(bpy)3]2+ in water and of the organization of its solvation shell in the low-spin and the high-spin states. In particular, the low-spin → high-spin change of states is shown to be accompanied (i) by a 0.191 Å lengthening of the Fe−N bond, in agreement with experiment, and (ii) by an increased thermal fluctuation of the molecular edifice, which both result from the weakening of the Fe−N bond. Furthermore, our results suggest that about two water molecules are expelled from the first solvation shell of [Fe(bpy)3]2+, which consists of water molecules intercalated between the bpy ligands. | ||||||||
|
||||||||
In the 3D network [Rh(bpy)3][NaCr(ox)3]ClO4 (ox = oxalate, bpy = 2,2'-bipyridine) phonon-assisted as well as resonant energy migration within the R1 line of the 4A2 → 2E transition of Cr3+ has been identified. The latter is dominant below 4.2 K, and in a fluorescence line narrowing spectrum, it manifests itself in a multiline pattern across the inhomogeneous line width with spacings corresponding to the zero-field splitting of the 4A2 ground state (Milos, M.; Kairouani, S.; Rabaste, S.; Hauser, A. Coord. Chem. Rev. 2008, 252, 2540). H. Riesen demonstrated efficient spectral hole burning within the R1 line of Cr3+ doped at low concentrations into partially deuterated NaMg[Al(ox)3]·9H2O (Riesen, H. Coord. Chem. Rev. 2006 250, 1737). Here we show that at higher Cr3+ concentrations in the same host, both phenomena can be observed simultaneously, the resonant energy migration thus creating an additional series of persistent side holes. | ||||||||
|
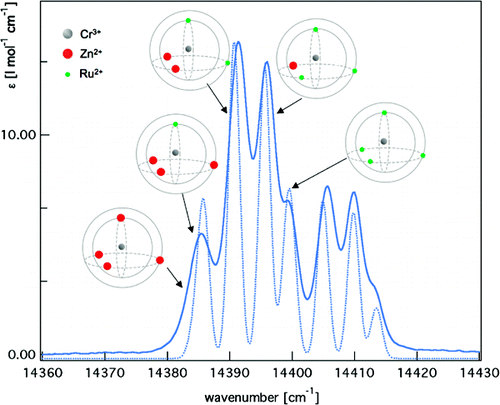 |
|||||||
In the mixed crystal series of the cubic three-dimensional networks of composition [Zn1−xRux(bpy)3][NaCr(ox)3] (0 ≤ x ≤1, ox = C2O42−, bpy = 2,2′-bipyridine), high-resolution absorption spectroscopy in the region of the 4A2→2E transition (R-lines) reveals the creation of five specific spectroscopic sites for the [Cr(ox)3]3− complex. The concentration of these spectroscopic sites follows a binomial distribution of [Zn(bpy)3]2+ and [Ru(bpy)3]2+ among the four nearest neighbors of a given [Cr(ox)3]3− complex within the network. The tris-bipyridine complexes occupying those positions have an optimal π−π interaction with the oxalate ligands of the tris-oxalate chromophore. The energy of each spectroscopic [Cr(ox)3]3− site depends on the total concentration of [Ru(bpy)3]2+ in the mixed crystal and on its specific distribution among the four nearest neighbors. Single crystal X-ray diffraction indicates a reduction of the unit cell volume when [Zn(bpy)3]2+ (a = 15.6365(18) Å) is substituted by [Ru(bpy)3]2+ (a = 15.5098(6) Å). This alone would lead to a red-shift of the R lines in analogy to the red-shift of 25.2 cm−1/GPa due to the decrease of the metal ligand Cr−O bond length as observed in high-pressure luminescence experiments. However, specific π−π interactions with the nearest neighbors have the opposite effect and shift the transition in discrete jumps to higher energies with increasing [Ru(bpy)3]2+ mole fraction. | ||||||||
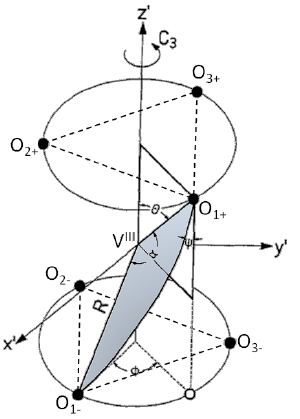 |
|
|||||||
The electronic structure and the photophysical properties of the vanadium(III)ion in pseudo-octahedral oxygen coordination is reviewed. V3+ has received much interest from spectroscopists in recent years due to the advancement of state-of-the-art experimental techniques such as inelastic neutron scattering and high-field electron paramagnetic resonance spectroscopy that directly interrogate its large ground state zero-field splittings (ZFSs) and to rational parameterization of the ligand fieldp arameters using the angular overlap model. However, for V3+ these ZFSs can be large enough to also be probed directly by high-resolution electronic absorption spectroscopy of intra-configurational (t22g → t22g) spin-forbidden transitions in the near-IR and visible regions. The luminescent properties of V3+ with hexa-oxo and tris-bidentate di-oxo-coordination are quite disappointing compared to its neighbor in the periodic table, Cr3+, in similar environments. The efficient non-radiative pathways in these compounds are reviewed and compared to recent work on V3+ doped into NaMgAl(ox)3⋅9H2O. The poor luminescence quantum efficiencies of V3+ oxo complexes is explained by strong coupling of multi-phonon processes with a dynamic Jahn-Teller distortion originating from the 3E trigonal component of the 3T1g ground state. | ||||||||
|
 |
|||||||
A molecular donor-acceptor dyad comprising a hexarhenium cluster core, [Re6(μ3-Se)8]2+, and a fullerene moiety which are covalently linked through a pyridine ligand was synthesized and fully characterized. The electrochemical and photophysical properties are reported. The detailed study includes cyclic voltammetry, steady-state absorption and fluorescence spectroscopy, radiation chemistry and transient absorption spectroscopy. A light-induced electron transfer between the inorganic cluster moiety and the fullerene can be excluded. However, a light-induced energy transfer from the rhenium cluster to the fullerene is proposed. | ||||||||
|
||||||||
The introduction of pump-probe techniques to the field of x-ray absorption spectroscopy (XAS) has allowed the monitoring of both structural and electronic dynamics of disordered systems in the condensed phase with unprecedented accuracy, both in time and in space. We present results on the electronically excited high-spin state structure of an Fe(II) molecular species, [FeII(bpy)3]2+, in aqueous solution, resolving the Fe-N bond distance elongation as 0.2 Å. In addition an analysis technique using the reduced χ2 goodness of fit between FEFF EXAFS simulations and the experimental transient absorption signal in energy space has been successfully tested as a function of excited state population and chemical shift, demonstrating its applicability in situations where the fractional excited state population cannot be determined through other measurements. Finally by using a novel ultrafast hard x-ray 'slicing' source the question of how the molecule relaxes after optical excitation has been successfully resolved using femtosecond XANES. | ||||||||
 |
|
|||||||
The ground-state electronic structures of K3V(ox)3·3H2O, Na3V(ox)3·5H2O, and NaMgAl1–xVx(ox)3·9H2O (0 < x <= 1, ox = C2O42–) have been studied by Fourier–transform electronic absorption and inelastic neutron scattering spectroscopies. High-resolution absorption spectra of the 3Γ(t2g2) → 1Γ(t2g2) spin-forbidden electronic origins and inelastic neutron scattering measurements of the pseudo-octahedral [V(ox)3]3– complex anion below 30 K exhibit both axial and rhombic components to the zero-field-splittings (ZFSs). Analysis of the ground-state ZFS using the conventional S = 1 spin Hamiltonian reveals that the axial ZFS component changes sign from positive values for K3V(ox)3·3H2O (D ≈ +5.3 cm–1) and Na3V(ox)3·5H2O (D ≈ +7.2 cm–1) to negative values for NaMgAl1–xVx(ox)3·9H2O (D ≈ –9.8 cm–1 for x = 0.013, and D ≈ –12.7 cm–1 for x = 1) with an additional rhombic component, |E|, that varies between 0.8 and 2 cm–1. On the basis of existing crystallographic data, this phenomenon can be identified as due to variations in the axial and rhombic ligand fields resulting from outer-sphere H-bonding between crystalline water molecules and the oxalate ligands. Spectroscopic evidence of a crystallographic phase change is also observed for K3V(ox)3·3Y2O (Y = H or D) with three distinct lattice sites below 30 K, each with a unique ground-state electronic structure. | ||||||||
|
 |
|||||||
The synthesis and structural characterization of a tetrathiafulvalene-fused perylenediimide molecular dyad is presented. Its largely extendedπ-conjugation provides intense optical absorption bands over a wide spectral range. The planar functional molecule exhibits a short-livednonluminescent excited state attributed to intramolecular charge separation. | ||||||||
|
||||||||
The relaxation in a spin transition compound is modeled on the basis of molecules interacting by theway of connecting springs and situated in a bidimensional open boundary hexagonal lattice. The switch ofindividual molecules is randomly checked using a standard Monte Carlo procedure. The switchingprobability depends on the energy gap between the two states in the absence of interactions and on theelongations of the nearest springs. The main characteristics of the experimental relaxation curves arereproduced and clustering and nucleation phenomena are detected. | ||||||||
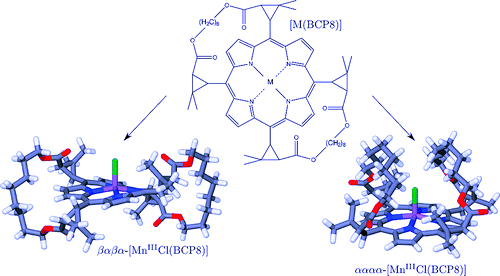 |
|
|||||||
Transition metal complexes of chiroporphyrins, in which two adjacent meso substituents are linked by a strap of eightmethylene groups, [M(BCP8)], can exist as either an αααα or αβαβ atropisomer depending on the nature of thecoordinated metal cation. This remarkable conformational versatility was investigated by density-functional theorycalculations for the d5 chloroiron(III) complex in the low-spin and high-spin states and for the d4 high-spinchloromanganese(III) complex. The lowest-lying electronic state of all of the conformers of the chloroiron(III) bridledchiroporphyrin is found to be the high-spin state. For the chloroiron(III) complex in the low-spin or the high-spin stateand for the high-spin chloromanganese(III) complex, the most stable form is predicted to be the αααα conformer inwhich the chloride axial ligand is located within the cavity provided by the bridles. The predicted stereochemistries arecompared with those similarly obtained (i) for the chloroiron(III) and chloromanganese(III) complexes of thetetramethylchiroporphyrin, which is devoid of straps, and (ii) for the d10 zinc(II) and low-spin d8 nickel(II) BCP8complexes, on the basis of the effects tied to the occupancy of the stereochemically active dx2-y2-type antibondingorbital level, to the restraints imposed by the straps, and to the presence of the axial chloride ligand. | ||||||||
|
||||||||
In the 3D oxalate networks [NaCr(ox)3][Rh(bpy)3]ClO4 and [NaCr(ox)3][Ru(bpy)3] (ox=oxalate, bpy=2,2′-bipyridine) three different types of energy migration within the 4A2→2E transition can be identified. One is a resonant process between spectral members spaced by the ground-state zero-field splitting (ZFS). This leads to the sequential appearance of additional sharp lines spaced by the ground-state ZFS in the fluorescence line narrowing spectrum across the inhomogeneous line. The second one is a quasi-resonant process between spectral neighbours and manifests itself by rapid spectral diffusion. The third one is the well-known phonon-assisted process setting in at higher temperature. | ||||||||
|
 |
|||||||
High-resolution Fourier transform absorption and luminescence spectroscopy reveal axial and rhombic zero-field splittings of the spin-forbidden electronic origins of V3+ in NaMgAl(ox)3·9H2O (ox=oxalate) single crystals below 25 K. The temperature dependence of the integrated absorption of the split features display behavior consistent with a Boltzmann distribution within the zero-field split 3Â2 ground state of V3+. Weak luminescence is observed in the near-IR from the lowest energy spin-forbidden transition with a luminescence lifetime of less than 0.5 μs at 11 K and an estimated quantum efficiency of the order of 10-5 | ||||||||
 |
|
|||||||
In order to study the electronic interactions in donor-acceptor ensembles as a function of pH, an efficient synthetic route to three imidazole-annulated tetrathiafulvalene (TTF) derivatives 1-3 is reported. Their electronic absorption spectra, in view of photoinduced intramolecular charge transfer, and their electrochemical behavior were investigated, and pKa values for the two protonation processes on the acceptor unit were determined in organic solvents by photometric titration. The influence of the TTF moiety on these values is discussed. | ||||||||
|
 |
|||||||
Triangular luminescent box: Self-assembly of a new multidentate receptor with europium cations results in the formation of trinuclear discrete complexes. X-ray crystallography shows that nine-coordinate cations are linked by ligands to provide a triangular complex in the solid state and in solution. Despite the coordinated solvent molecules, this topologically unusual complex exhibits remarkable luminescent properties. | ||||||||
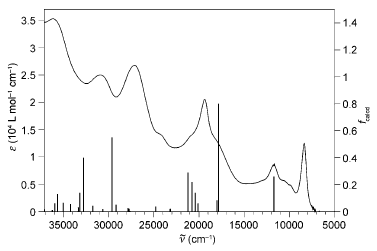 |
|
|||||||
Electronic absorption spectrum of 1 in DMF solution at room temperature, together with the calculated oscillator strengths. | ||||||||
|
 |
|||||||
Density functional theory is applied within a supramolecular approach to the study of the guest−host interactions in [Fe(bpy)3]2+@Y and their influence on the structural, energetic, and 57Fe Mössbauer spectroscopy properties of the encapsulated [Fe(bpy)3]2+ complex in the low- and high-spin states. The structures of the isolated complex and the supramolecular model used for [Fe(bpy)3]2+@Y were optimized in both spin-states using different generalized gradient approximation (PBE, HCTH, OLYP) and hybrid (B3LYP*, O3LYP) functionals. The results obtained are consistent with one another and show that, in either spin-state, the structure of [Fe(bpy)3]2+ shrinks and distorts upon encapsulation. Still, the structural changes experienced by the complex in a given spin-state remain limited, especially in that they do not lead to a substantial variation of the 57Fe quadrupole splitting, whose calculated values are in very good agreement with avalaible experimental data. The decomposition of the guest−host interaction energy into its electrostatic, Pauli and orbital contributions shows that the bonding between the complex and the supercage is more electrostatic than covalent. The ability of modern functionals to accurately describe the interactions explains the remarkable consistency of the results obtained with the various functionals. In particular, although the functionals perform very differently for the determination of the high-spin/low-spin energy difference ΔEHLel in [Fe(bpy)3]2+ and [Fe(bpy)3]2+@Y, they consistently predict that the encapsulation entails a destabilization of the high-spin state with regard to the low-spin state of Δ(ΔEHLel) = 2500 cm−1. Using for [Fe(bpy)3]2+ the CASPT2 value of ΔEHLel = 3700 cm−1 [Pierloot, K.; Vancoillie, S. J. Chem. Phys.2006, 125, 124303; Pierloot, K.; Vancoillie, S. J. Chem. Phys.2008, 128, 034104], we obtain for the high-spin/low-spin energy difference in [Fe(bpy)3]2+@Y, a best ab initio estimate of ΔEHLel = 6200 cm−1. | ||||||||
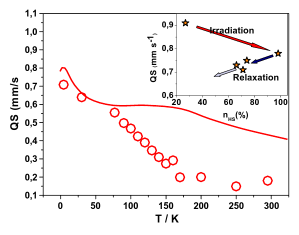 |
|
|||||||
The spin-transition (1A1↔5T2) behaviour of a new mononuclear iron(II) compound [FeII(L)3][PF6]2[L = 2-[3-(2′-pyridyl)pyrazole-1-ylmethyl]pyridine] has been investigated by 57Fe Mössbauer spectroscopy. Analysis of the Mössbauer spectra revealed low value of the quadrupole splitting of the high-spin state which reflects iron(II) to be in nearly cubic lattice site. Mössbauer spectra under light show the light-induced excited spin state trapping effect and the observed quadrupole splitting of the metastable high-spin state is found little sensitive to the high-spin fraction value. DFT calculations are in progress to document the almost cubic nature of the ligand-field acting on the iron atom. | ||||||||
|
||||||||
The trigonal planar geometry of the nitrogen atom in commonly used phosphoramidite ligands is not in line with the traditional valence shell electron pair repulsion (VSEPR) model. In this work, the effects governing nitrogen configuration in several substituted aminophosphines, A2PNB2 (A or B = H, F, Cl, Br, Me, OMe, BINOP), are examined using modern computational analytic tools. The electron delocalization descriptions provided by both electron localization function (ELF) and block localized wavefunction analysis support the proposed relationships between conformation and negative hyperconjugative interactions. In the parent H2PNH2, the pyramidal nitrogen configuration results from nitrogen lone pair electron donation into the σ* P — H orbital. While enhanced effects are seen for F2PNMe2, placing highly electronegative fluorine substituents on nitrogen (i.e., Me2PNF2) eliminates delocalization of the nitrogen lone pair. Understanding and quantifying these effects can lead to greater flexibility in designing new catalysts. | ||||||||
|
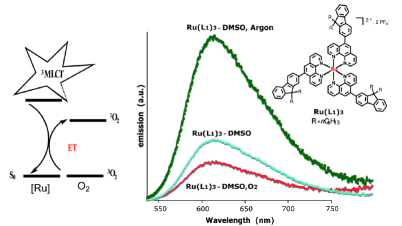 |
|||||||
The synthesis and characterization of new 1,10-phenanthroline-based chromophores LT1, LT2 and LD1 featuring fluorene unit(s) are reported. Their absorption and emission as well as their two-photon absorption properties in the 450–650 nm spectral range are discussed in comparison with the parent 1,10-phenanthroline and already described ligands L1 and L2. | ||||||||
 |
|
|||||||
A π-extended, redox-active bridging ligand 4′,5′-bis(propylthio)tetrathiafulvenyl[i]dipyrido[2,3-a:3′,2′-c]phenazine (L) was prepared via direct Schiff-base condensation of the corresponding diamine−tetrathiafulvalene (TTF) precursor with 4,7-phenanthroline-5,6-dione. Reactions of L with [Ru(bpy)2Cl2] afforded its stable mono- and dinuclear ruthenium(II) complexes 1 and 2. They have been fully characterized, and their photophysical and electrochemical properties are reported together with those of [Ru(bpy)2(ppb)]2+ and [Ru(bpy)2(μ-ppb)Ru(bpy)2]4+ (ppb = dipyrido[2,3-a:3′,2′-c]phenazine) for comparison. In all cases, the first excited state corresponds to an intramolecular TTF → ppb charge-transfer state. Both ruthenium(II) complexes show two strong and well-separated metal-to-ligand charge-transfer (MLCT) absorption bands, whereas the 3MLCT luminescence is strongly quenched via electron transfer from the TTF subunit. Clearly, the transient absorption spectra illustrate the role of the TTF fragment as an electron donor, which induces a triplet intraligand charge-transfer state (3ILCT) with lifetimes of approximately 200 and 50 ns for mono- and dinuclear ruthenium(II) complexes, respectively. | ||||||||
 |
||||||||
The mononuclear OsII complex [Os( L1)3](PF6)2 ( L1 = 5-methyl(1-methylbenzimidazol-2-yl)pyridine) is an obvious candidate for the design of an inert d-block-based tripodal receptor capable of binding and photosensitizing trivalent lanthanides (LnIII). It has thus been prepared and its two enantiomeric meridional (Δ-mer and Λ-mer) and facial (rac-fac) isomers have been separated by ion-exchange chromatography. The optical isomers have been characterized by CD spectroscopy and assignments of absolute configuration confirmed by an X-ray crystallographic study of Λ-mer-[Os( L1)3](PF6)2·1.5MeCN (monoclinic, P21, Z = 4). Comparison of the latter structure with that of racemic fac-[Os( L1)3](PF6)2 (monoclinic, C2/c, Z = 8) and [Os(bipy)3](PF6)2 (where bipy = 2,2' -bipyridine) shows minimal structural variations, but differences are observed in the photophysical and electrochemical properties of the respective compounds. Luminescence emissions from OsII complexes of L1 are typically lower in energy, with shorter lifetimes and lower quantum yields than their bipy analogues, whilst metal-centred oxidation processes are more facile due to the enhanced π-donor ability of L1. The key relationships between these parameters are discussed. Finally, though challenged by (i) the low reactivity of many osmium precursors and (ii) the irreversible formation of competing side products, the synthesis and purification of the heterobimetallic triple-stranded helicate HHH-[OsLu( L2)3](CF3SO3)5 has been realised, in which L2 is a segmental ligand containing the same bidentate unit as that found in L1 further connected to a tridentate binding site adapted for complexing LnIII. Its solid-state structure has been established by X-ray crystallography (triclinic, P1-, Z = 2). | ||||||||
|
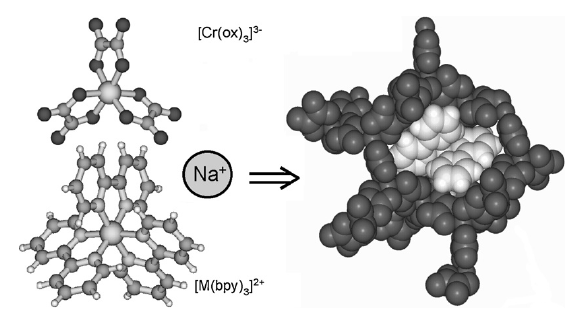 |
|||||||
Excitation energy migration is an important phenomenon at high concentration of luminescent chromophores. In crystalline solids it results in a quenching of the intrinsic luminescence of the chromophore as the excitation energy migrates to impurity centres or other forms of trap sites. As concluded from the extensively studied systems where Cr3+ is doped as the active chromophore into inert host lattices, energy migration in crystalline solids is usually a phonon-assisted process, in which the simultaneous creation or annihilation of phonons helps to bridge the energy miss-match in the energy levels of two neighbouring chromophores within a inhomogeneously broadened absorption band. However, in the three-dimensional network systems [Ru(bpy)3][NaCr(ox)3] and [Rh(bpy)3][NaCr(ox)3]ClO4, it proved possible to unambiguously identify three different mechanisms for energy migration within the R1 line of the 4A2 → 2E transition of Cr3+. In addition to the common temperature dependant phonon-assisted process, a resonant process between the zero-field split components of the 4A2 ground state leading to a multi-line pattern in a fluorescence line narrowing spectrum and a quasi-resonant process within the same component leading to fast spectral diffusion can be identified at very low temperature. The parameters governing these processes are discussed and the behaviour of the model systems is compared to more conventional doped oxides and related systems. | ||||||||
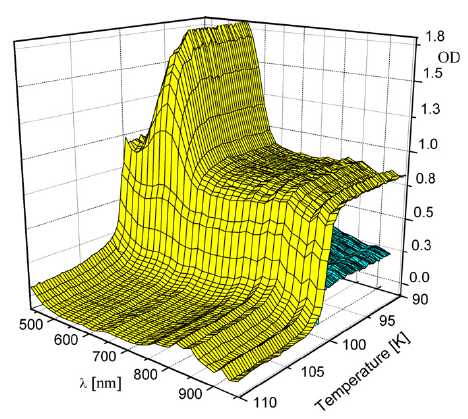 |
|
|||||||
The thermal and the light-induced spin transition in [Fe(bbtr)3](ClO4)2 (bbtr = 1,4-di(1,2,3-triazol-1-yl)) as well as the high-spin → low-spin relaxation following the light-induced population of the high-spin state below the thermal transition temperature are discussed in relation to the accompanying crystallographic phase transition. The experimental data have exclusively been obtained using optical single crystal absorption spectroscopy. | ||||||||
|
||||||||
The spin-crossover compound [Fe(bbtr)3](ClO4)2 (bbtr = 1,4-di(1,2,3-triazol-1-yl)butane) forms a polymeric hexagonal sheet structure. It shows an abrupt thermal spin transition with 13 K wide hysteresis around 105 K, as evidenced by single crystal optical spectroscopy. The transition temperature for the thermal high-spin→low-spin transition on cooling as well as the relaxation kinetics just below Tc↓ depend upon the history of the sample. This is typical for a nucleation and growth mechanism and domain formation. In contrast, the high-spin→low-spin relaxation following the light-induced population of the high-spin state at low temperatures is governed by the intersystem crossing process. | ||||||||
|
||||||||
Two new ethynylbipyridine-linked mono- and bis-tetrathiafulvalene (TTF) derivatives, together with a Ru(II) complex, were synthesized using Sonogashira coupling reactions and characterized by UV/vis spectroscopy and cyclic voltammetry. They display a clear electrochemically amphoteric behavior consisting of two reversible single-electron oxidation waves (typical for TTF derivatives) and one reversible single-electron reduction wave (bpy) and act as donor–acceptor (D–A) systems. Furthermore, for the Ru(II) complex, a quite intense fluorescence originating from the 3MLCT state is observed. | ||||||||
|
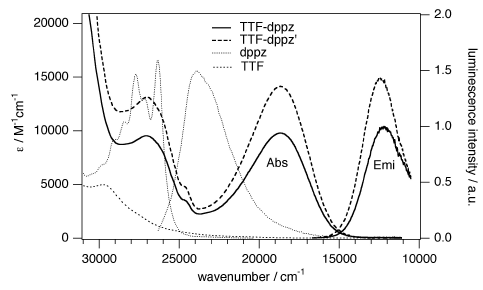 |
|||||||
The creation of long-lived charge-separated states in donor-acceptor assemblies has been the goal of many studies aimed at mimicking the primary processes in photosynthesis. Here we present such assemblies based on tetrathiafulvalene (TTF) as electron donor and a dipyridophenazine (dppz) unit as electron acceptor in the form of a fused ligand (TTF-dppz) coordinated to ruthenium(II) via the dipyrido coordination site and with 2,2′-bipyridine (bpy) as auxiliary ligand, namely [Ru(bpy)3−x(TTF-dppz)x]2+ (x = 1−3). For x = 2, irradiation into the metal to dppz charge transfer transition results in electron transfer from TTF to ruthenium, thus creating a charge-separated state best described by [(TTF+-dppz)Ru(dppz−-TTF)(bpy)]2+ with a lifetime of 2.5 μs in dichloromethane. | ||||||||
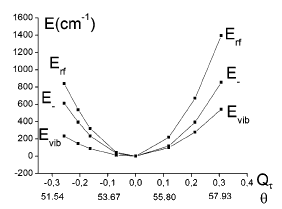 |
|
|||||||
The topology of the ground-state potential energy surface of M(CN)6 with orbitally degenerate 2T2g (M = TiIII (t2g1), FeIII and MnII (both low-spin t2g5)) and 3T1g ground states (M = VIII (t2g2), MnIII and CrII (both low-spin t2g4)) has been studied with linear and quadratic Jahn−Teller coupling models in the five-dimensional space of the εg and τ2g octahedral vibrations (Tg⊗(εg+τ2g) Jahn−Teller coupling problem (Tg = 2T2g, 3T1g)). A procedure is proposed to give access to all vibronic coupling parameters from geometry optimization with density functional theory (DFT) and the energies of a restricted number of Slater determinants, derived from electron replacements within the t2g1,5 or t2g2,4 ground-state electronic configurations. The results show that coupling to the τ2g bending mode is dominant and leads to a stabilization of D3d structures (absolute minima on the ground-state potential energy surface) for all complexes considered, except for [Ti(CN)6]3-, where the minimum is of D4h symmetry. The Jahn−Teller stabilization energies for the D3d minima are found to increase in the order of increasing CN−M π back-donation (TiIII < VIII < MnIII < FeIII < MnII < CrII). With the angular overlap model and bonding parameters derived from angular distortions, which correspond to the stable D3d minima, the effect of configuration interaction and spin−orbit coupling on the ground-state potential energy surface is explored. This approach is used to correlate Jahn−Teller distortion parameters with structures from X-ray diffraction data. Jahn−Teller coupling to trigonal modes is also used to reinterpret the anisotropy of magnetic susceptibilities and g tensors of [Fe(CN)6]3-, and the 3T1g ground-state splitting of [Mn(CN)6]3-, deduced from near-IR spectra. The implications of the pseudo Jahn−Teller coupling due to t2g−eg orbital mixing via the trigonal modes (τ2g) and the effect of the dynamic Jahn−Teller coupling on the magnetic susceptibilities and g tensors of [Fe(CN)6]3- are also addressed. | ||||||||
|
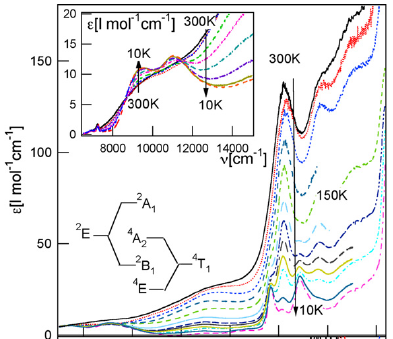 |
|||||||
The spin transition of the [Co(terpy)2]2+ complex (terpy = 2,2′:6′,2″-terpyridine) is analysed based on experimental data from optical spectroscopy and magnetic susceptibility measurements. The single crystal absorption spectrum of [Co(terpy)2](ClO4)2 shows an asymmetric absorption band at 14 400 cm−1 with an intensity typical for a spin-allowed d–d transition and a temperature behaviour typical for a thermal spin transition. The single crystal absorption spectra of suggest that in this compound, the complex is essentially in the high-spin state at all temperatures. However, the increase in intensity observed in the region of the low-spin MLCT transition with increasing temperature implies an unusual partial thermal population of the low-spin state of up to about 10% at room temperature. Finally, high-spin → low-spin relaxation curves following pulsed laser excitation for [Co(terpy)2](ClO4)2 dispersed in KBr discs, and as a comparison for the closely related [Co(4-terpyridone)2](ClO4)2 spin-crossover compound are given. | ||||||||
|
||||||||
An unsymmetric, peripherally octasubstituted phthalocyanine (Pc) 1, which contains a combination of dipyrido[3,2-f:2‘,3‘-h] quinoxaline and 3,5-di-tert-butylphenoxy substituents, has been obtained via a statistical condensation reaction of two corresponding phthalonitriles. Synthetic procedures for the selective metalation of the macrocyclic cavity and the periphery of 1 were developed, leading to the preparation of the key precursor metallophthalocyanines 3−5 in good yields. Two different strategies were applied to the synthesis of compact dyads MPc−Ru(II) 6−8 (M = Mg(II), Co(II), Zn(II)). Intramolecular electronic interactions in these dyads were studied by absorption, emission, and transient absorption spectroscopy. Upon photoexcitation, these dyads exhibit efficient intramolecular energy transfer from the Ru(II) chromophore to the MPc moiety. | ||||||||
|
||||||||
The influence of pressure on the structural and vibrational properties of a2RuH6has been investigated using periodic density functional theory calculations performed at the local density approximation (LDA) and generalized gradient approximation (GGA) levels. At ambient pressure, the calculated structure and vibrational frequencies are in satisfactory agreement with experimental data. The calculated em>P-Vcurves could be fitted with the Vinet equation of state, yielding em>B0=67.6and em>B0=58.5 GPaat the LDA and GGA levels, respectively, and em>B0′=4.0at both theoretical levels. The unit cell parameter is found to decrease faster with increasing pressure than the Ru–H bond length. The calculated pressure dependence of the vibrational frequencies agrees well with experiment for em>ν5(T2g)but not for em>ν9(A1g) | ||||||||
|
||||||||
Three ruthenium(II) polypyridine complexes of general formula [Ru(bpy)3-n(TTF-dppz)n](PF6)2 (n=1-3, bpy=2,2'-bipyridine), with one, two or three redox-active TTF-dppz (4',5'-bis(propylthio)tetrathiafulvenyl[i]dipyrido[3,2-a:2',3'-c]phenazine) ligands, were synthesised and fully characterised. Their electrochemical and photophysical properties are reported together with those of the reference compounds [Ru(bpy)3](PF6)2, [Ru(dppz)3](PF6)2 and [Ru(bpy)2(dppz)](PF6)2 and the free TTF-dppz ligand. All three complexes show intraligand charge-transfer (ILCT) fluorescence of the TTF-dppz ligand. Remarkably, the complex with n=1 exhibits luminescence from the Ru2+dppz metal-to-ligand charge-transfer (3MLCT) state, whereas for the other two complexes, a radiationless pathway via electron transfer from a second TTF-dppz ligand quenches the 3MLCT luminescence. The TTF fragments as electron donors thus induce a ligand-to-ligand charge-separated (LLCS) state of the form TTF-dppz--Ru2+→ -dppz-TTF+. The lifetime of this LLCS state is approximately 2.3 μs, which is four orders of magnitude longer than that of 0.4 ns for the ILCT state, because recombination of charges on two different ligands is substantially slower. | ||||||||
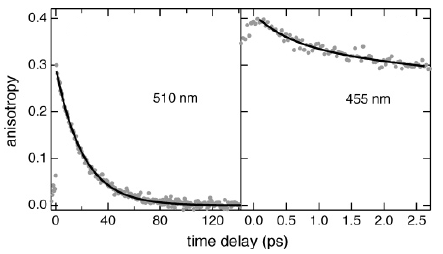 |
|
|||||||
The influence of solute−solvent interactions on the vibrational energy relaxation dynamics of perylene and substituted perylenes in the first singlet excited-state upon excitation with moderate (<0.4 eV) excess energy has been investigated by monitoring the early narrowing of their fluorescence spectrum. This narrowing was found to occur on timescales ranging from a few hundreds of femtoseconds to a few picoseconds. Other processes, such as a partial decay of the fluorescence anisotropy and the damping of a low-frequency oscillation due to the propagation of a vibrational wavepacket, were found to take place on a very similar time scale. No significant relationship between the strength of nonspecific solute−solvent interactions and the vibrational energy relaxation dynamics of the solutes could be evidenced. On the other hand, in alcohols the spectral narrowing is faster with a solute having H-bonding sites, indicating that this specific interaction tends to favor vibrational energy relaxation. No relationship between the dynamics of spectral narrowing and macroscopic solvent properties, such as the thermal diffusivity, could be found. On the other hand, a correlation between this narrowing dynamics and the number of low-frequency modes of the solvent molecules was evidenced. All these observations cannot be discussed with a model where vibrational energy relaxation occurs via two consecutive and dynamically well-separated steps, namely ultrafast intramolecular vibrational redistribution followed by slower vibrational cooling. On the contrary, the results indicate that both intra- and intermolecular vibrational energy redistribution processes are closely entangled and occur, at least partially, on similar timescales. | ||||||||
|
||||||||
We describe an advanced setup for time-resolved x-ray absorption fine structure (XAFS) Spectroscopy with picosecond temporal resolution. It combines an intense femtosecond laser source synchronized to the x-ray pulses delivered into the microXAS beamline of the Swiss Light Source (SLS). The setup is applied to measure the short-lived high-spin geometric structure of photoexcited aqueous Fe(bpy)3 at room temperature. | ||||||||
|
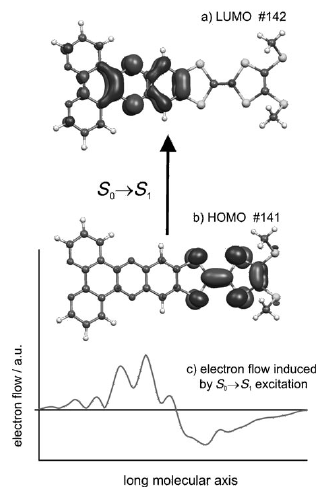 |
|||||||
To study the electronic interactions in donor-acceptor (D-A) ensembles, D and A fragments are coupled in a single molecule. Specifically, a tetrathiafulvalene (TTF)-fused dipyrido[3,2-a:2',3'-c]phenazine (dppz) compound having inherent redox centers has been synthesized and structurally characterized. Its electronic absorption, fluorescence emission, photoinduced intramolecular charge transfer, and electrochemical behavior have been investigated. The observed electronic properties are explained on the basis of density functional theory. | ||||||||
|
||||||||
We report an in-depth theoretical study of 4-styrylpyridine in its singlet S0 ground state. The geometries and the relative stabilities of the trans and cis isomers were investigated within density functional theory (DFT) as well as within Hartree-Fock (HF), second-order Møller-Plesset (MP2), and coupled cluster (CC) theories. The DFT calculations were performed using the B3LYP and PBE functionals, with basis sets of different qualities, and gave results that are very consistent with each other. The molecular structure is thus predicted to be planar at the energy minimum, which is associated with the trans conformation, and to become markedly twisted at the minimum of higher energy, which is associated with the cis conformation. The results of the calculations performed with the post-HF methods approach those obtained with the DFT methods, provided that the level of treatment of the electronic correlation is high enough and that sufficiently flexible basis sets are used. Calculations carried out within DFT also allowed the determination of the geometry and the energy of the molecule at the biradicaloid transition state associated with the thermal cis ↔trans isomerization and at the transition states associated with the enantiomerization of the cis isomer and with the rotations of the pyridinyl and phenyl groups in the trans and cis isomers. Car-Parrinello molecular dynamics simulations were also performed at 50, 150, and 300 K using the PBE functional. The studies allowed us to evidence the highly flexible nature of the molecule in both conformations. In particular, the trans isomer was found to exist mainly in a nonplanar form at finite temperatures, while the rotation of the pyridinyl ring in the cis isomer was incidentally observed to take place within ≈1 ps during the simulation carried out at 150 K on this isomer. | ||||||||
|
||||||||
Structural changes of the iron(II)-tris-bipyridine ([FeII(bpy)3]2+) complex induced by ultrashort pulse excitation and population of its short-lived (≤0.6 ns) quintet high spin state have been detected by picosecond x-ray absorption spectroscopy. The structural relaxation from the high spin to the low spin state was followed over the entire lifetime of the excited state. A combined analysis of the x-ray-absorption near-edge structure and extended x-ray-absorption fine structure spectroscopy features delivers an Fe-N bond elongation of 0.2 Å in the quintet state compared to the singlet ground state. | ||||||||
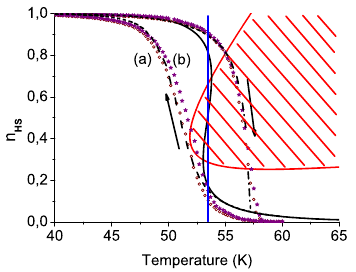 |
|
|||||||
We present novel insight on like-spin domains (LSD) in cooperative spin transition solids by following the photo-transformation and the subsequent relaxation of a [Fe(ptz)6](BF4)2 single crystal in the vicinity of the light-induced instability. Self-organization under light is observed, accompanied by Barkhausen-like noise and jumps which reveal the presence of elastic interactions between LSDs. The light-induced phase separation process is discussed in terms of a dynamic potential providing spinodal instability in the corresponding temperature range. This useful concept is applicable to all types of switchable molecular solids. | ||||||||
|
||||||||
Whereas there are hundreds of known iron(II) spin-crossover compounds, only a handful of cobalt(II) spin-crossover compounds have been discovered to date, and hardly an in depth study on any of them exists. This review begins with an introduction into the theoretical aspects to be considered when discussing spin-crossover compounds in general and cobalt(II) systems in particular. It is followed by case studies on [Co(bpy)3]2+ and [Co(terpy)2]2+ (bpy = 2,2′-bipyridine, terpy = 2,2′:6′,2″-terpyridine) presenting and discussing results from magnetic susceptibility measurements, X-ray crystallography, optical spectroscopy, and EPR spectroscopy. | ||||||||
|
||||||||
Circular dichroism (CD) spectra and density functional theory (DFT) calculations are reported for a series of conformationally bistable chiroporphyrins with 8-methylene bridles MBCP-8, which can display either an αααα or an αβαβ orientation of their meso substituents. From DFT geometry optimizations, the most stable form of ZnBCP-8 is found to be the αααα conformer. By passing to NiBCP-8, there is a strong stabilization of the αβαβ conformation with respect to the αααα conformation, consistent with the X-ray structures of αααα-ZnBCP-8 and αβαβ-NiBCP-8. A correlation between the sign of the CD signal in the Soret region and the conformation of the BCP-8 compounds is reported: the αααα conformers H2BCP-8 and ZnBCP-8 show a positive CD signal, whereas the αβαβ conformers NiBCP-8 and CuBCP-8 exhibit a negative signal. The possible contributions to the rotational strengths of αβαβ-NiBCP-8 and αααα-ZnBCP-8, calculated on the basis of their crystal structures, have been analyzed. The CD signals are found to result from a combination of both the inherent chirality of the porphyrin and of extrinsic contributions due to the chiral bridles. These results may have a broad significance for understanding the chiroptical properties of chiral porphyrins and hemoproteins and for monitoring stimuli-responsive, conformationally bistable chiroporphyrin compounds. | ||||||||
|
 |
|||||||
Photoswitching of the dielectric constant has been observed for the first time in the spin-crossover complex [Fe(L)(CN)2]·H2O (L=2,13-dimethyl-6,9-dioxa-3,12,18-triazabicyclo[12.3.1]octadeca-1(18),2,12,14,16-pentaene, see picture). The electrical detection of a photoinduced change in spin state could allow the use of such complexes in optical information-storage devices. | ||||||||
|
||||||||
Frequency shifts of the Ag I 4d105s 2S1∕2(F=0,MF=0) to 4d95s2 2D5∕2(F′=2,MF′=0) electric-quadrupole transition at 330.6 nm due to external fields are calculated using multiconfigurational self-consistent field methods. As this forbidden transition is free from first order Doppler and Zeeman effects, it is under investigation for the realization of an atomic optical clock. The calculated perturbations are the light shift, the blackbody frequency shift, and the quadratic Zeeman shift. Results show that a total uncertainty of 10−18 could be reach without confining the atoms in a Lamb-Dicke regime in an optical lattice. | ||||||||
|
||||||||
A series of molybdenum and tungsten nitrido, [M(N)(X)(diphos)2], and imido complexes, [M(NH)(X)(diphos)2)]Y, (M = Mo, W) with diphosphine coligands (diphos = dppe/depe), various trans ligands (X = N3-, Cl-, NCCH3) and different counterions (Y- = Cl-, BPh4-) is investigated. These compounds are studied by infrared and Raman spectroscopies; they are also studied with isotope-substitution and optical-absorption, as well as emission, spectroscopies. In the nitrido complexes with trans-azido and -chloro coligands, the metal−N stretch is found at about 980 cm-1; upon protonation, it is lowered to about 920 cm-1. The 1A1 → 1E (n → π*) electronic transition is observed for [Mo(N)(N3)(depe)2] at 398 nm and shows a progression in the metal−N stretch of 810 cm-1. The corresponding 3E → 1A (π* → n) emission band is observed at 542 nm, exhibiting a progression in the metal−N stretch of 980 cm-1. In the imido system [Mo(NH)(N3)(depe)2]BPh4, the n → π* transition is shifted to lower energy (518 nm) and markedly decreases in intensity. In the trans-nitrile complex [Mo(N)(NCCH3)(dppe)2]BPh4, the metal−N(nitrido) stretching frequency increases to 1016 cm-1. The n → π* transition now is found at 450 nm, shifting to 525 nm upon protonation. Most importantly, the reduction of this nitrido trans-nitrile complex is drastically facilitated compared to its counterparts with anionic trans-ligands (Epred = −1.5 V vs Fc+/Fc). On the other hand, the basicity of the nitrido group is decreased (pKa{[Mo(NH)(NCCH3)(dppe)2](BPh4)2} = 5). The implications of these findings with respect to the Chatt cycle are discussed. | ||||||||
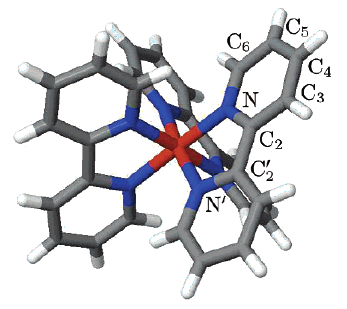 |
|
|||||||
State-of-the-art generalized gradient approximation (GGA) (PBE, OPBE, RPBE, OLYP, and HCTH), meta-GGA (VSXC and TPSS), and hybrid (B3LYP, B3LYP*, O3LYP, and PBE0) functionals are compared for the determination of the structure and the energetics of the D3 [Co(bpy)3]2+ complex in the 4A2 and 4E trigonal components of the high-spin 4T1g( t52g e2g ) state and in the low-spin 2E state of octahedral 2Eg( t62g e1g) parentage. Their comparison extends also to the investigation of the Jahn−Teller instability of the 2E state through the characterization of the extrema of C2 symmetry of this spin state's potential energy surface. The results obtained for [Co(bpy)3]2+ in either spin manifold are very consistent among the functionals used and are in good agreement with available experimental data. The functionals, however, perform very differently with respect to the spin-state energetics because the calculated values of the high-spin/low-spin energy difference ΔEelHL vary between −3212 and 3919 cm-1. Semilocal functionals tend to give too large ΔEelHL values and thus fail to correctly predict the high-spin state as the ground state of the isolated complex, while hybrid functionals tend to overestimate the stability of the high-spin state with respect to the low-spin state. Reliable results are, however, obtained with the OLYP, HCTH, B3LYP*, and O3LYP functionals which perform best for the description of the isolated complex. The optical properties of [Co(bpy)3]2+ in the two spin states are also analyzed on the basis of electronic excitation calculations performed within time-dependent density functional response theory. The calculated absorption and circular dichroism spectra agree well with experimental results. | ||||||||
|
||||||||
A new 1,3-dithiol-2-ylidene substituted naphthopyranone 2 has been synthesized and characterized. UV–vis spectroscopic and cyclic voltammetry results, interpreted on the basis of density functional theory, show that 2 displays an intramolecular charge-transfer transition and acts like a donor–acceptor (D–A) system. Furthermore, a weak fluorescence originating from the excited charge-transfer state is observed. | ||||||||
|
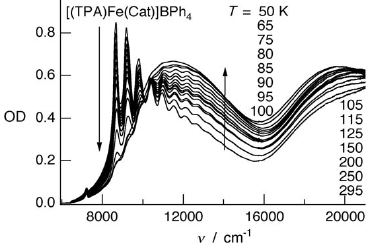 |
|||||||
The photophysical properties of the ferric catecholate spin-crossover compounds [(TPA)Fe(R-Cat)]X (TPA=tris(2-pyridylmethyl)amine; X=PF6-, BPh4-; R-Cat=catecholate dianion substituted by R=NO2, Cl, or H) are investigated in the solid state. The catecholate-to-iron(III) charge-transfer bands are sensitive both to the spin state of the metal ion and the charge-transfer interactions associated with the different catecholate substituents. Vibronic progressions are identified in the near-infrared (NIR) absorption of the low-spin species. Evidence for a low-temperature photoexcitation process is provided. The relaxation dynamics between 10 and 100 K indicate a pure tunneling process below ≈40 K, and a thermally activated region at higher temperatures. The relaxation rate constants in the tunneling regime at low temperature, kHL(T→0), vary in the range from 0.58 to 8.84 s-1. These values are in qualitative agreement with the inverse energy-gap law and with structural parameters. A comparison with ferrous spin-crossover complexes shows that the high-spin to low-spin relaxation is generally faster for ferric complexes, owing to the smaller bond length changes for the latter. However, in the present case the corresponding rate constants are smaller than expected based on the single configurational coordinate model. This is attributed to the combined influence of the electronic configuration and the molecular geometry. | ||||||||
|
||||||||
The high-spin → low-spin relaxation in spin-crossover compounds can be described as non-adiabatic multi-phonon process in the strong coupling limit, in which the low-temperature tunnelling rate increases exponentially with the zero-point energy difference between the two states. Based on the hypothesis that the experimental bond length difference between the high-spin and the low-spin state of ~0.2 Å is also valid for low-spin iron(II) complexes, extrapolation of the single configurational coordinate model allows an estimate of the zero-point energy difference for low-spin complexes from kinetic data. DFT calculations on low-spin [Fe(bpy)3]2+ support the structural assumption. However, for low-spin [Fe(terpy)2]2+ the relaxation rate constant shows an anomalous behaviour in so far as it is more in line with spin-crossover systems. This is attributed to very anisotropic bond length changes associated with the spin state change, and the subsequent breakdown of the single mode model. | ||||||||
|
||||||||
A planar π-conjugated heteroaromatic molecule 1 has been synthesized and fully characterized; it combines two characteristics, a charge-transfer transition originating from its inherent donor–acceptor nature in its neutral state and an intervalence charge-transfer transition in its 12+ mixed-valence state. | ||||||||
|
||||||||
In supramolecular systems, the interaction between different units modulates their photophysical properties. a) For platinum(II) complexes with ligands that have extended π systems, π-stacking and direct metal-metal interactions result in the formation of excimers with characteristically red-shifted luminescence. Time-resolved emission spectra show clear evidence of dual luminescence. b) In phthalocyanines to which electron-donating tetrathiafulvalene (TTF) groups have been fused, the luminescence is strongly quenched by intramolecular electron transfer. The luminescence can be switched on by oxidation of the TTF groups. c) The luminescence of ruthenium tris-bipyridyl derivatives is strongly influenced by the environment. Linked to biotin, the luminescence quantum yield of such a complex is enhanced by 30 % upon binding to avidin. Furthermore, the binding to avidin induces a circular-dichroism signal from the π-π* transition of the initially racemic ruthenium tris-bipyridyl derivative. | ||||||||
|
||||||||
The molecular structure of the highly oxygen-sensitive complex [L3CuI(NCCH3)](BF4) (1) reveals approximately symmetrical coordination by the fac-tridentate (tripodal) ligand L3 = tris(3-isopropyl-4,5-trimethylenepyrazolyl)methane and a rather short CuI-N(acetonitrile) distance of 1.865(5) Å. In CH2Cl2 at -78 °C the colourless compound reacts with O2 to yield a labile purple intermediate (λmax 517 nm) - presumably a peroxodicopper(II) complex - which decomposes at -30 °C. No such intermediate was observed on reaction of the CuI complex of bis(2-pyridylmethyl)benzylamine with O2 at -80 °C. However, an EPR spectrum with g|| = 2.17 and g_|_ = 2.03 without 63,65Cu hyperfine splitting was observed at low temperatures. Exposure of the precursor 1 to air under ambient conditions yields dinuclear [L3Cu||(μ-OH)2CuIIL3](BF4)2 (2) which exhibits an EPR detectable dissociation into monomers in CH2Cl2 solution. The structure of the hexakis(dichloromethane) solvate of 2 with Cu-Cu and Cu-O distances of 3.055 and 1.94Å, respectively, is typical for dihydroxo-bridged dicopper compounds with square-pyramidal Cu|| configuration (τ = 0.03), adopting an anti arrangement. In agreement with the relatively wide Cu-O-Cu angles of 103.5° an analysis of the temperature dependence of the magnetic susceptibility revealed a rather strong (J = -633 cm-1) antiparallel spin-spin coupling. The effect is ascribed to the steric bulk of the ligand L3. | ||||||||
|
||||||||
The optical properties of a thin film of the [Ru(bpy)3][NaCr(ox)3] network structure obtained by pulsed laser deposition are described. The luminescence shows the characteristic doublet of R lines at 14 400 cm−1 of the spin-forbidden ligand field transition 2E(t2g3)→4A2(t2g3) of the [Cr(ox)3]3− chromophore. The resonant energy migration within the R1 line shows that the three-dimensional crystallographic structure is preserved during the coating process. The observation of the R lines of [Cr(bpy)3]3+ at 13 710 cm−1 indicates that a small fraction of Cr3+ ions migrate from the oxalate network to the tris-bipyridine cation site in the cavities of the network. | ||||||||
 |
|
|||||||
The cubic Prussian blue analogue Mn3[Mn(CN)6]2 · 15 H2O, which has the advantage of being transparent and magnetic (TN = 35 K) at the same time, has been investigated by density functional theory (DFT) calculations. The three-dimensional structure is built of MnII ions linked to MnIII ions by μ-bridging cyanides, to form a crystal structure, which is related to the NaCl type. In a first step, the relative stabilities of the mononuclear complexes [Mn(CN)6]z- (z = 2 to 4) have been studied as a function of the oxidation state, spin configuration, and the linkage isomerism of the cyanide ligand. The results we have obtained by this investigation are in good agreement with our chemical expertise. In addition, the calculations have been extended to the dinuclear [Mn2(CN)11]z- (z = 5 and 6) clusters. Furthermore, we used DFT to model the magnetic properties as well as the 3T1 → 1T2 transition, which has been observed by single-crystal near-IR spectra of Mn3[Mn(CN)6]2 · 15 H2O. | ||||||||
|
||||||||
The compound {Fe(pmd)[Ag(CN)2][Ag2(CN)3]} (pmd=pyrimidine) was synthesized and characterized. Magnetic, calorimetric and single crystal visible spectroscopic studies demonstrate the occurrence of a two-step high-spin (HS) ↔ low-spin (LS) transition. The critical temperatures are Tc1=185 and Tc2=148 K. Each step involves ~50 % of the iron centers, with the low-temperature step showing a hysteresis of 2.5 K. The enthalpy and entropy variations associated with the two steps are ΔH1=3.6±0.4 kJ mol-1 and ΔS1=19.5±3 J K-1 mol-1; ΔH2=4.8±0.4 kJ mol-1 and ΔS2=33.5±3 J K-1 mol-1. Photomagnetic and visible spectroscopy experiments show that below 50 K, where the LS state is the thermodynamically stable state, the compound can be switched quantitatively to the HS state using green-red light (550-650 nm). HS-to-LS relaxation experiments in the dark at temperatures between 15 and 55 K show that the relaxation takes place via a two-step cooperative process, which was analyzed in the context of the mean field theory. The crystal structure has been studied at 290, 220, 170, 90 and 30 K together with 30 K after irradiation. The compound adopts monoclinic symmetry (P21/c, Z=16) at all temperatures. There are five [FeN6] pseudo-octahedral sites linked by pmd, [Ag(CN)2]- and [Ag2(CN)3]- bridging ligands to form an unprecedented three-dimensional (6,6) topology. The structural analysis allows for an understanding of the microscopic mechanism of the two-step behavior of the thermally induced spin transition as well as the corresponding relaxation of the photoexcited compound based on the individual changes of the five sites. Synergy between metallophilic interactions and the spin transition is also shown by the variation of the Ag…Ag distances. Correlations between the variation of the unit-cell volume and the change of Ag…Ag interactions within each step with the asymmetric change of the anomalous heat capacity have also been inferred. | ||||||||
|
||||||||
The ability of different density functionals to describe the structural and energy differences between the high- [5T2g:(t2g)4(eg)2] and low- [1A1g:(t2g)6(eg)0] spin states of small octahedral ferrous compounds is studied. This work is an extension of our previous study of the hexaquoferrous cation, [Fe(H2O)6]2+, [J. Chem. Phys. 120, 9473 (2004)] to include a second compound—namely, the hexaminoferrous cation, [Fe(NH3)6]2+—and several additional functionals. In particular, the present study includes the highly parametrized generalized gradient approximations (GGAs) known as HCTH and the meta-GGA VSXC [which together we refer to as highly parametrized density functionals (HPDFs)], now readily available in the GAUSSIAN03 program, as well as the hybrid functional PBE0. Since there are very few experimental results for these molecules with which to compare, comparison is made with best estimates obtained from second-order perturbation theory-corrected complete active space self-consistent field (CASPT2) calculations, with spectroscopy oriented configuration interaction (SORCI) calculations, and with ligand field theory (LFT) estimations. While CASPT2 and SORCI are among the most reliable ab initio methods available for this type of problem, LFT embodies many decades of empirical experience. These three methods are found to give coherent results and provide best estimates of the adiabatic low-spin–high-spin energy difference, ΔELHadia, of 12 000–13 000 cm−1 for [Fe(H2O)6]2+ and 9 000–11 000 cm−1 for [Fe(NH3)6]2+. All functionals beyond the purely local approximation produce reasonably good geometries, so long as adequate basis sets are used. In contrast, the energy splitting, ΔELHadia, is much more sensitive to the choice of functional. The local density approximation severely over stabilizes the low-spin state with respect to the high-spin state. This “density functional theory (DFT) spin pairing-energy problem” persists, but is reduced, for traditional GGAs. In contrast the hybrid functional B3LYP underestimates ΔELHadia by a few thousands of wave numbers. The RPBE GGA of Hammer, Hansen, and Nørskov gives good results for ΔELHadia as do the HPDFs, especially the VSXC functional. Surprisingly the HCTH functionals actually over correct the DFT spin pairing-energy problem, destabilizing the low-spin state relative to the high-spin state. Best agreement is found for the hybrid functional PBE0. | ||||||||
|
||||||||
Inert and optically active pseudo-octahedral CrIIIN6 and RuIIN6 chromophores have been incorporated by self-assembly into heterobimetallic triple-stranded helicates HHH-[CrLnL3]6+ and HHH-[RuLnL3]5+. The crystal structures of [CrLnL3](CF3SO3)6 (Ln=Nd, Eu, Yb, Lu) and [RuLnL3](CF3SO3)5 (Ln=Eu, Lu) demonstrate that the helical structure can accommodate metal ions of different sizes, without sizeable change in the intermetallic M…Ln distances. These systems are ideally suited for unravelling the molecular factors affecting the intermetallic nd→4f communication. Visible irradiation of the CrIIIN6 and RuIIN6 chromophores in HHH-[MLnL3]5/6+ (Ln=Nd, Yb, Er; M=Cr, Ru) eventually produces lanthanide-based near infrared (NIR) emission, after directional energy migration within the complexes. Depending on the kinetic regime associated with each specific d-f pair, the NIR luminescence decay times can be tuned from micro- to milliseconds. The origin of this effect, together with its rational control for programming optical functions in discrete heterobimetallic entities, are discussed. | ||||||||
|
 |
|||||||
In the iron(II) low-spin complex [Fe(bpy)3]2+, the zero-point energy difference between the 5T2g(t42ge2g) high-spin and the 1A1g(t62g) low-spin states, ΔE0HL, is estimated to lie in the range of 2500-5000 cm-1. This estimate is based on the low-temperature dynamics of the high-spin→low-spin relaxation following the light-induced population of the high-spin state and on the assumption that the bond-length difference between the two states ΔrHL is equal to the average value of ≈0.2 Å, as found experimentally for the spin-crossover system. Calculations based on density functional theory (DFT) validate the structural assumption insofar as the low-spin-state optimised geometries are found to be in very good agreement with the experimental X-ray structure of the complex and the predicted high-spin geometries are all very close to one another for a whole series of common GGA (PB86, PW91, PBE, RPBE) and hybrid (B3LYP, B3LYP*, PBE1PBE) functionals. This confirmation of the structural assumption underlying the estimation of ΔE0HL from experimental relaxation rate constants permits us to use this value to assess the ability of the density functionals for the calculation of the energy difference between the HS and LS states. Since the different functionals give values from -1000 to 12000 cm-1, the comparison of the calculated values with the experimental estimate thus provides a stringent criterion for the performance of a given functional. Based on this comparison the RPBE and B3LYP* functionals give the best agreement with experiment. | ||||||||
|
||||||||
The synthesis of tetrakis(tetrathiafulvalene)-annulated metal-free and metallophthalocyanines 5−8 via the tetramerization of the phthalonitrile derivative 4 is reported. All of them have been fully characterized by electronic absorption spectroscopy, thin-layer cyclic voltammetry, mass spectrometry, and elemental analysis. Their solution electrochemical data show two reversible four-electron oxidation waves, indicating that these fused systems are strong π-electron donors, which give rise to tetra- or octaradical cation species. For the metal-free phthalocyanine 5, additionally a reversible one-electron wave was found in the negative direction arising from the reduction of the macrocycle. Moreover, the tetrathiafulvalene unit acts as an efficient reductive electron-transfer quencher for the phthalocyanine emission, but upon its oxidation, an intense luminescence is switched on. | ||||||||
|
||||||||
Visible pump−probe spectroscopy has been used to identify and characterize short-lived metal-to-metal charge transfer (MMCT) excited states in a group of cyano-bridged mixed-valence complexes of the formula [LCoIIINCMII(CN)5]-, where L is a pentadentate macrocyclic pentaamine (L14) or triamine-dithiaether (L14S) and M is Fe or Ru. Nanosecond pump−probe spectroscopy on frozen solutions of [L14CoIIINCFeII(CN)5]- and [L14SCoIIINCFeII(CN)5]- at 11 K enabled the construction of difference transient absorption spectra that featured a rise in absorbance in the region of 350−400 nm consistent with the generation of the ferricyanide chromophore of the photoexcited complex. The MMCT excited state of the Ru analogue [L14CoIIINCRuII(CN)5]- was too short-lived to allow its detection. Femtosecond pump−probe spectroscopy on aqueous solutions of [L14CoIIINCFeII(CN)5]- and [L14SCoIIINCFeII(CN)5]- at room temperature enabled the lifetimes of their CoII−FeIII MMCT excited states to be determined as 0.8 and 1.3 ps, respectively. | ||||||||
|
||||||||
Excitation energy transfer processes play an important role in many areas of physics, chemistry and biology. The three-dimensional oxalate networks of composition [MIII(bpy)3][MIMIII(ox)3]ClO4 (bpy=2,2-bipyridine, ox=oxalate, MI=alkali ion) allow for a variety of combinations of different transition metal ions. The combination with chromium(III) on both the tris-bipyridine as well as the tris-oxalate site constitutes a model system in which it is possible to differentiate unambiguously between energy transfer from [Cr(ox)3]3– to [Cr(bpy)3]3+ due to dipole-dipole interaction on the one hand and exchange interaction on the other hand. Furthermore it is possible to just as unambiguously differentiate between the common temperature dependent phonon-assisted energy migration within the 2E state of [Cr(ox)3]3–, and a unique resonant process. | ||||||||
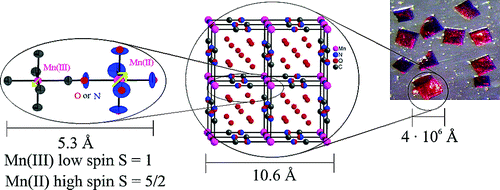 |
|
|||||||
The compound of stoichiometry Mn(II)3[Mn(III)(CN)6]2·zH2O (z = 12−16) (1) forms air-stable, transparent red crystals. Low-temperature single crystal optical spectroscopy and single crystal X-ray diffraction provide compelling evidence for N-bonded high-spin manganese(II), and C-bonded low-spin manganese(III) ions arranged in a disordered, face-centered cubic lattice analogous to that of Prussian Blue. X-ray and neutron diffraction show structured diffuse scattering indicative of partially correlated (rather than random) substitutions of [Mn(III)(CN)6] ions by (H2O)6 clusters. Magnetic susceptibility measurements and elastic neutron scattering experiments indicate a ferrimagnetic structure below the critical temperature Tc = 35.5 K. | ||||||||
|
||||||||
The phenomenon of the thermal spin transition, as observed for octahedral transition metal complexes having a d 4 to d 7 electronic configuration, can be fully rationalised on the basis of ligand field theory. In order to arrive at a self-consistent description of the vibronic structure of spin crossover compounds, it is essential to take into account the fact that the population of anti-bonding orbitals in the high-spin state results in a substantially larger metal-ligand bond length than for the low-spin state. Whereas the electron-electron repulsion is not affected to any great extent by such a bond length difference, the ligand field strength for iron(II) spin crossover compounds can be estimated to be almost twice as large in the low-spin state as compared to the one for the high-spin state. In fact, the dependence of the ligand field strength on the metal-ligand distance may be considered the quantum mechanical driving force for the spin crossover phenomenon. | ||||||||
|
||||||||
The discovery of a light-induced spin transition at cryogenic temperatures in a series of iron(II) spin-crossover compounds in 1984 has had an enormous impact on spin-crossover research. Apart from being an interesting photophysical phenomenon in its own right, it provided the means of studying the dynamics of the intersystem crossing process between the high-spin and the low-spin state in a series of compounds and over a large temperature range. It could thus be firmly established that intersystem crossing in spin-crossover compounds is a tunnelling process, with a limiting low-temperature lifetime below 50 K and a thermally activated region above 100 K. This review begins with an elucidation of the mechanism of the light-induced spin transition, followed by an in depth discussion of the chemical and physical factors, including cooperative effects, governing the lifetimes of the light-induced metastable states. | ||||||||
|
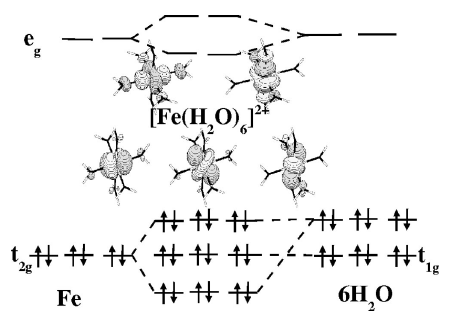 |
|||||||
A comparison of density functionals is made for the calculation of energy and geometry differences for the high- [5T2g: (t2g)4(eg)2] and low- [1A1g: (t2g)6(eg)0] spin states of the hexaquoferrous cation [Fe(H2O)6]2+. Since very little experimental results are available (except for crystal structures involving the cation in its high-spin state), the primary comparison is with our own complete active-space self-consistent field (CASSCF), second-order perturbation theory-corrected complete active-space self-consistent field (CASPT2), and spectroscopy-oriented configuration interaction (SORCI) calculations. We find that generalized gradient approximations (GGAs) and the B3LYP hybrid functional provide geometries in good agreement with experiment and with our CASSCF calculations provided sufficiently extended basis sets are used (i.e., polarization functions on the iron and polarization and diffuse functions on the water molecules). In contrast, CASPT2 calculations of the low-spin–high-spin energy difference ΔELH = ELS−EHS appear to be significantly overestimated due to basis set limitations in the sense that the energy difference of the atomic asymptotes (5D→1I excitation of Fe2+) are overestimated by about 3000 cm−1. An empirical shift of the molecular ΔELH based upon atomic calculations provides a best estimate of 12 000–13 000 cm−1. Our unshifted SORCI result is 13 300 cm−1, consistent with previous comparisons between SORCI and experimental excitation energies which suggest that no such empirical shift is needed in conjunction with this method. In contrast, after estimation of incomplete basis set effects, GGAs with one exception underestimate this value by 3000–4000 cm−1 while the B3LYP functional underestimates it by only about 1000 cm−1. The exception is the GGA functional RPBE which appears to perform as well as or better than the B3LYP functional for the properties studied here. In order to obtain a best estimate of the molecular ΔELH within the context of density functional theory (DFT) calculations we have also performed atomic excitation energy calculations using the multiplet sum method. These atomic DFT calculations suggest that no empirical correction is needed for the DFT calculations. | ||||||||
|
||||||||
In the three-dimensional oxalate network structures of composition [CoxMII1-x(bpy)3][MICr(ox)3], the spin state of the [Cox(bpy)3]2+ complex can be tuned by means of chemical pressure. With MI=Na it is a classic high-spin complex. Substitution of Na by Li stabilises the complex and it becomes a spin-crossover complex. Dilution with MII=Fe reinforces this effect, and MII=Zn reverses it. | ||||||||
|
||||||||
Unsymmetrical substituted bidentate benzimidazol-2-ylpyridine ligands L2 and L3 react with [Ru(dmso)4Cl2] in ethanol to give statistical 1:3 mixtures of fac-[Ru(Li)3]2+ and mer-[Ru(Li)3]2+ (i=2, 3; ΔGΘisomerisation=-2.7 kJ mol-1). In more polar solvents (acetonitrile, methanol), the free energy of the facial ↔ meridional isomerisation process favours mer-[Ru(Li)3]2+, which is the only isomer observed in solution at the equilibrium (ΔGΘisomerisation≤-11.4 kJ mol-1). Since the latter process takes several days for [Ru(L2)3]2+, fac-[Ru(L2)3]2+ and mer-[Ru(L2)3]2+ have been separated by chromatography, but the 28-fold increase in velocity observed for [Ru(L3)3]2+ provides only mer-[Ru(L3)3](ClO4)2 after chromatography (RuC60H51N9O8Cl2, monoclinic, P21/n, Z=4). The facial isomer can be stabilised when an appended tridentate binding unit, connected at the 5-position of the benzimidazol-2-ylpyridine unit in ligand L1, interacts with nine-coordinate lanthanides(III). The free energy of the facial↔meridional isomerisation is reversed (ΔGΘisomerisation≥11.4 kJ mol-1), and the Ru — N bonds are labile enough to allow the quantitative thermodynamic self-assembly of HHH-[RuLu(L1)3]5+ within hours ([RuLu(L1)3](CF3SO3)4.5Cl0.5(CH3OH)2.5: RuLuC106H109Cl0.5N21O19S4.5F13.5, triclinic, P, Z=2). Electrochemical and photophysical studies show that the benzimidazol-2-ylpyridine units in L1-L3 display similar π-acceptor properties to, but stronger π-donor properties than, those found in 2,2'-bipyridine. This shifts the intraligand π→π* and the MLCT transitions toward lower energies in the pseudo-octahedral [Ru(Li)3]2+ (i=2, 3) chromophores. The concomitant short lifetime of the 3MLCT excited state points to efficient, thermally activated quenching via low-energy Ru-centred d-d states, a limitation which is partially overcome by mechanical coupling in HHH-[RuLu(L1)3]5+. | ||||||||
|
||||||||
Magnetization measurements and variable temperature optical spectroscopy have been used to investigate, within the 4−300 K temperature range, the electronic structure of the reduced high-potential iron protein (HiPIP) from Chromatium vinosum and the model compounds (Cat)2[Fe4S4(SR)4], where RS- = 2,4,6-triisopropylphenylthiolate (1), 2,6-diphenylphenylthiolate (2), diphenylmethylthiolate (3), 2,4,6-triisopropylbenzylthiolate (4, 4‘), 2,4,6-triphenylbenzylthiolate (5, 5‘), 2,4,6-tri-tert-butylbenzylthiolate (6), and Cat+ = +NEt4 (1, 2, 3, 4‘, 5‘, 6), +PPh4 (4, 5). The newly synthesized 22-, 32-, 52-, and 62- complexes are, as 12- and 42-, excellent models of the reduced HiPIPs: they exhibit the [Fe4S4]3+/2+ redox couple, because of the presence of bulky ligands which stabilize the [Fe4S4]3+ oxidized core. Moreover, the presence of SCH2 groups in 42-, 52-, and 62-, as in the [Fe4S4] protein cores, makes them good biomimetic models of the HiPIPs. The X-ray structure of 2 is reported: it crystallizes in the orthorhombic space group Pcca with no imposed symmetry and a D2d-distorted geometry of the [Fe4S4]2+ core. Fit of the magnetization data of the reduced HiPIP and of the 1, 2, 3, 4, 5, and 6 compounds within the exchange and double exchange theoretical framework leads to exchange coupling parameters J = 261−397 cm-1. A firm determination of the double exchange parameters B or, equivalently, the transfer integrals β = 5B could not be achieved that way. The obtained |B| values remain however high, attesting thus to the strength of the spin-dependent electronic delocalization which is responsible for lowest lying electronic states being characterized by delocalized mixed-valence pairs of maximum spin 9/2. Electronic properties of these systems are then accounted for by the population of a diamagnetic ground level and excited paramagnetic triplet and quintet levels, which are respectively J and 3J above the ground level. Optical studies of 1, 2, 4‘, 5‘, and 6 but also of (NEt4)2[Fe4S4(SCH2C6H5)4] and the isomorph (NEt4)2[Fe4S4(S-t-Bu)4] and (NEt4)2[Fe4Se4(S-t-Bu)4] compounds reveal two absorption bands in the near infrared region, at 705−760 nm and 1270−1430 nm, which appear to be characteristic of valence-delocalized and ferromagnetically coupled [Fe2X2]+ (X = S, Se) units. The |B| and |β| values can be directly determined from the location at 10|B| of the low-energy band, and are respectively of 699−787 and 3497−3937 cm-1. Both absorption bands are also present in the 77 K spectrum of the reduced HiPIP, at 700 and 1040 nm (Cerdonio, M.; Wang, R.-H.; Rawlings, J.; Gray, H. B. J. Am. Chem. Soc. 1974, 96, 6534−6535). The blue shift of the low-energy band is attributed to the inequivalent environments of the Fe sites in the protein, rather than to an increase of |β| when going from the models to the HiPIP. The small differences observed in known geometries of [Fe4S4]2+ clusters, especially in the Fe−Fe distances, cannot probably lead to drastic changes in the direct Fe−Fe interactions (parameter β) responsible for the delocalization phenomenon. These differences are however magnetostructurally significant as shown by the 261−397 cm-1 range spanned by J. The cluster's geometry, hence the efficiency of the Fe−μ3-S−Fe superexchange pathways, is proposed to be controlled by the more or less tight fit of the cluster within the cavity provided by its environment. | ||||||||
|
|
 |
|
|||||||
Copper(II) complexes of the pentadentate bispidine ligands exist in two isomeric forms (see structure) with bond-length differences up to 0.5 Å. The stabilization of either isomer may be achieved by a variation of the substituent at N7. | ||||||||
|
||||||||
In this paper we discuss on the quantum efficiency in spin crossover compounds. Spin crossover solids are text-book examples of photo switchable materials that present a thermal spin transition from the diamagnetic low-spin state, thermodynamically stable at low temperatures, to the paramagnetic high-spin state becoming the thermodynamically stable state at elevated temperature. By irradiating them with an appropriate wavelength, they can pass from the stable low spin state to the metastable high spin state at temperatures below the thermal transition temperature. For the compound [Fe(pic)3]Cl2·EtOH, the question regarding the quantum efficiency of the photo-conversion process that is the number of molecules converted by one single photon and its possible dependency on irradiation intensity gave rise to a controversy. The experimental results presented in this paper demonstrate that the quantum efficiency of the photo-conversion at 11 K is on the order of unity, with no noticeable dependency of the quantum efficiency on light intensity. It does, however, depend to a small extent on the fraction of complexes already converted to the high-spin state. | ||||||||
|
||||||||
The relation between the internal pressure during spin-crossover is compared to the chemical pressure induced by dilution with zinc. Further, the light of a specific LIESST (Light Induced Excited Spin State Trapping) wavelength is used to induce partial stabilisation of high-spin state and thus shift temperature of the spin-crossover towards lower values. The de-coupling of the spin-crossover and structural phase transition is discussed. | ||||||||
|
||||||||
A novel ruthenium complex with a 6,7-dicyanosubstituted dppz ligand has been synthesised: its crystal structure and physico-chemical studies are reported. | ||||||||
|
||||||||
The physical and photophysical properties of three classic transition metal complexes, namely [Fe(bpy)3]2+, [Ru(bpy)3]2+, and [Co(bpy)3]2+, can be tuned by doping them into a variety of inert crystalline host lattices. The underlying guest-host interactions are discussed in terms of a chemical pressure. | ||||||||
|
||||||||
The [Ru(bpy)3][LiCr(ox)3] system (bpy = 2,2‘-bipyridine, ox = oxalate) has two crystallographically non-equivalent [Cr(ox)3]3- sites. In steady-state resonant and nonresonant fluorescence line narrowing (FLN) experiments on the R1 lines of the two non-equivalent [Cr(ox)3]3- chromophores, multiline spectra are observed at 1.6 K. Such multiline spectra are clear evidence for resonant energy transfer processes within the inhomogeneously broadened R1 lines. In addition, time-resolved experiments show that also site-to-site energy transfer occurs, which turns out to be resonant, too, however with a non-negligible phonon-assisted contribution even at 1.5 K. | ||||||||
|
||||||||
Efficient resonant energy transfer occurs within the R1 line of the 4A2 → 2E transition of the [Cr(ox)3]3- chromophore in mixed crystal [Rh(bpy)3][NaAl1-xCrx(ox)3]ClO4 (x = 0.05−0.9, ox = oxalate, bpy = 2,2‘-bipyridine). This manifests itself in the form of multiline patterns in resonant fluorescence line narrowing (FLN) experiments at 1.5 K. The conditions for such a resonant process to occur are that the inhomogeneous line width of the R1 line is larger than the zero-field splitting of the ground state, which, in turn, is larger than the homogeneous line width of the transition. The number of lines and their relative intensities depend critically upon the [Cr(ox)3]3- concentration and the excitation wavelength within the inhomogeneous distribution. The basic model for resonant energy transfer as presented by von Arx et al. (Phys. Rev B 1996, 54, 15800) is extended to include the effects of diluting the chromophores in an inert host lattice and of nonresonant R2 excitation. In addition, Monte Carlo simulations serve to explain the temporal evolution of the multiline pattern following pulsed excitation. | ||||||||
|
 |
|||||||
[Fe(pic)3]Cl2·EtOH (pic = 2-picolylamine) is a spin-crossover compound that can be converted from the low-spin state to the high-spin state at temperatures below the thermal transition temperature by way of light irradiation in the visible part of the electromagnetic spectrum. For this compound, the question regarding the quantum efficiency of this photoconversion process and its possible dependence on irradiation intensity gave rise to some controversy. The experimental results presented in this paper demonstrate that the quantum efficiency of the photoconversion at 11 K is on the order of unity, with no noticeable dependence on irradiation intensity. It does, however, depend to some extent on the fraction of complexes already converted to the high-spin state. | ||||||||
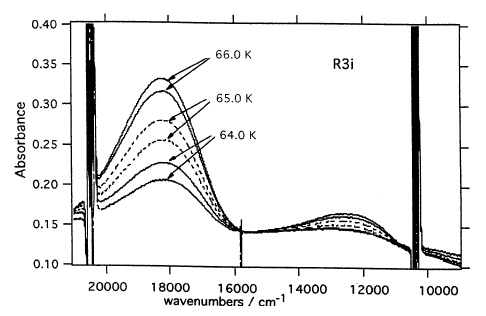 |
|
|||||||
The quasi-static nature of a light induced thermal hysteresis was studied on the spin-transition compound [Fe(ptz)6](BF4)2, by means of optical spectroscopy and magnetic measurements in the temperature interval between 10 and 80 K. Various experimental procedures are discussed in relation to the competition between the two processes considered, namely the photoexitation and the high-spin→low-spin relaxation. A detailed discussion of the experimental parameters, which should be considered in order to avoid erroneous interpretations of LITH, is given. | ||||||||
|
||||||||
The light-induced high-spin → low-spin relaxation for the Fe(II) spin-crossover compounds [Fe(btpa)](PF6)2 and [Fe(b(bdpa))](PF6)2 in solution, where btpa is the potentially octadentate ligand N,N,N‘,N‘-tetrakis(2-pyridylmethyl)-6,6‘-bis(aminomethyl)-2,2‘-bipyridine and b(bdpa) is the analogous hexadentate ligand N,N‘-bis(benzyl)-N,N‘-bis(2-pyridylmethyl)-6,6‘-bis(aminomethyl)-2,2‘-bipyridine, respectively, has been studied by temperature-dependent laser flash photolysis. [Fe(b(bdpa))](PF6)2 shows single-exponential 5T2 → 1A1 relaxation kinetics, whereas [Fe(btpa)](PF6)2 exhibits solvent-independent biphasic relaxation kinetics. The fast process of [Fe(btpa)](PF6)2 with a rate constant, kHL, of 2.5 × 107 s-1 at 295 K and an activation energy, Ea, of 1294(26) cm-1 in methanol can be assigned to the 5T2 → 1A1relaxation as well. The slow process with a kHL(295 K) of 3.7 × 105 s-1 and a Ea of 2297(32) cm-1 in methanol - which is the slowest light-induced relaxation process observed so far for an Fe(II) spin-crossover complex in solution - is assigned to a coupling of the 5T2 → 1A1relaxation process to a geometrical rearrangement within the pendent pyridyl arms. | ||||||||
|
||||||||
Present paper is an overview of our efforts during the past few years to understand complicated corelations of physical phenomena related to pressure in Fe(I1) solid state spin transition systems. Some principal results concerning p, T, λ-experiments are extracted. In the context of correlation of the crystallographic phase transition with simultaneous HS → LS relaxation and LS → HS photopopulation, we show the latest results: Brillouin and magnetic measurements on the crystal [Fe(pt6](BF6)2. | ||||||||
|
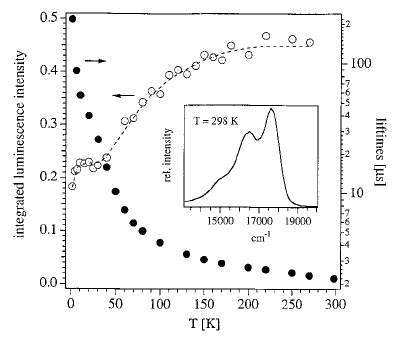 |
|||||||
Luminescence and energy transfer in [Zn1-xRux(bpy)3][NaAl1-yCry(ox)3] (x ≈ 0.01, y = 0.006 − 0.22; bpy = 2,2‘-bipyridine, ox = C2O42-) and [Zn1-x-yRuxOsy(bpy)3][NaAl(ox)3] (x ≈ 0.01, y = 0.012) are presented and discussed. Surprisingly, the luminescence of the isolated luminophores [Ru(bpy)3]2+ and [Os(bpy)3]2+ in [Zn(bpy)3][NaAl(ox)3] is hardly quenched at room temperature. Steady-state luminescence spectra and decay curves show that energy transfer occurs between [Ru(bpy)3]2+ and [Cr(ox)3]3- and between [Ru(bpy)3]2+ and [Os(bpy)3]2+ in [Zn1-xRux(bpy)3][NaAl1-yCry(ox)3] and [Zn1-x-yRuxOsy(bpy)3] [NaAl(ox)3], respectively. For a quantitative investigation of the energy transfer, a shell type model is developed, using a Monte Carlo procedure and the structural parameters of the systems. A good description of the experimental data is obtained assuming electric dipole−electric dipole interaction between donors and acceptors, with a critical distance Rc for [Ru(bpy)3]2+ to [Cr(ox)3]3- energy transfer of 15 Å and for [Ru(bpy)3]2+ to [Os(bpy)3]2+ energy transfer of 33 Å. These values are in good agreement with those derived using the Förster−Dexter theory. | ||||||||
|
||||||||
Incorporation of [Co(bpy)3]2+ into the cavities of the three-dimensional oxalate network structure in [Co(bpy)3][LiCr(ox)3] produces chemical pressure that destabilises the normal high-spin ground state 4T1 to such an extent that the [Co(bpy)3]2+ complex becomes a spin-crossover complex. It shows a temperature-dependent equilibrium between the 2E low-spin and the 4T1 high-spin states. | ||||||||
|
||||||||
Short range correlations of the distribution of high spin (HS) and low spin (LS) states show up in thermal spin transition curves, decay curves of the light induced metastable HS state (LIESST state), and in structural features during the spin transitions. Correlations are due to short range interactions between the spin crossover molecules. Short range interactions may compete with omnipresent long range interactions and give rise to interesting spin transition phenomena. In this paper, the effect of correlations on the thermal spin transition in the mixed crystal system [FexZn1−x(pic)3]Cl2·EtOH (pic=picolylamine) is discussed. In particular the step in the thermal transition curve is a direct consequence of such correlations. In addition, the decay of the metastable HS state of the pure iron compound at ca. 20 K can be significantly changed by preparing metastable HS states with a random distribution over the lattice sites. Both experiments could be well reproduced by Monte Carlo simulations. In the orthorhombic modification of the compound Fe[5NO2-sal-N(1,4,7,10)]([2,2′-(2,5,8,11-tetraazadodeca-1,11-diene-1,12-diyl)4-nitrophenolato] (2-)-N2, N2′,N2′′,N2′′′,O1, O1′]Fe(II)) a commensurable superstructure has been found. This compound represents the first example of a stable infinite range correlation of the spin states over the lattice sites. | ||||||||
|
||||||||
The propagation of the high-spin (HS) → low-spin (LS) relaxation at 53 K in a single crystal of the iron (II) spin-crossover compound [Fe(ptz)6](BF4)2 was followed by photography, after inducing the local photoexcitation to the metastable HS state at 20 K using the single wavelength (457 nm Ar± ion laser) irradiation. The photoinduced formation of the HS—LS patterns with a characteristic diameter of some 0.1 mm was observed to occur inhomogeneously at a macroscopic scale already during photoexcitation. The contrast between the HS (transparent) and the LS (purple) regions was amplified during relaxation. The effect is described in terms of a transient instability, for which a microscopic model in the mean-field approximation is proposed. The mechanism for the development of patterns at the macroscopic scale is discussed. | ||||||||
|
||||||||
Electronic energy transfer from [Cr(ox)3]3- (ox = oxalate) in three-dimensional (3D) anionic oxalate networks to encapsulated [Cr(bpy)3]3+ (bpy = 2,2‘-bipyridine) cations at 1.5 K was investigated by time-resolved luminescence spectroscopy. Two series of mixed crystals of nominal compositions [NaAl1-xCrx(ox)3][Rh0.99Cr0.01(bpy)3]ClO4 (x = 0, 0.01, 0.05, 0.1, 0.2, 0.4, 0.6, 0.8, and 1) and [NaAl0.99Cr0.01(ox)3][Rh1-yCry(bpy)3]ClO4 (y = 0, 0.01, 0.02, 0.03, 0.04, and 0.05) were utilized. Energy transfer from [Cr(ox)3]3- to [Cr(bpy)3]3+ occurs by two mechanisms. Rapid, short-range transfer (ket > 106 s-1) is attributed to superexchange coupling between the Cr3+ ions via π overlap of the oxalate and bipyridine ligands. In addition, at low [Cr(ox)3]3- concentrations (nominally x = 0.01) a very much slower process with a maximum ket ≈ 200 s-1 is identified in the time-resolved spectra and attributed to a dipole−dipole mechanism. Furthermore, the resonant [Cr(ox)3]3- to [Cr(ox)3]3- energy migration previously reported by von Arx et al. (Phys. Rev. (1996), B54, 15800) assists [Cr(ox)3]3- to [Cr(bpy)3]3+ transfer as the [Cr(ox)3]3- concentration increases. | ||||||||
 |
|
|||||||
In iron(II) spin-crossover compounds, the transition from the 1A1 low-spin state at low temperatures to the 5T2 high-spin state at elevated temperatures is accompanied by a large increase in metal-ligand bond lengths. The resulting elastic interactions may be pictured as an internal pressure which is proportional to the concentration of the low-spin species. Because pressure stabilises the low-spin state relative to the high-spin state this results in a positive feedback. Thermal transition curves in neat iron(II) spin-crossover compounds are thus invariable much steeper than in diluted mixed crystals, and the high-spin→low-spin relaxation following the light-induced population of the high-spin state at low temperatures is self-accelerating. Strong interactions give rise to a thermal hysteresis, and light-induced bistabilities may be observed for compounds with initially a high-spin ground state and the potential for a light-induced population of the low-spin state. For such compounds, the increasing internal pressure may stabilise the low-spin state sufficiently so that it becomes the molecular ground state above some critical light-induced low-spin fraction. Secondary effects of the elastic interactions include crystallographic phase transitions, inhomogeneous distributions of sites, and anomalies such as steps in the transition curve. | ||||||||
|
|
||||||||
Temperature-dependent laser flash photolysis experiments on the low-spin iron(II) systems [M1−xFex(bpy)3](PF6)2 (M=Cd, Mn and Zn,x≈0.01, bpy=2,2′-bipyridine) under external pressure are presented. Below 50 K the high-spin→low-spin relaxation is an almost temperature-independent tunnelling process. Above that temperature it tends towards a thermally activated behaviour. A change of the host from cadmium to zinc results in an increase of the low-temperature tunnelling rate constant by two orders of magnitude. An external pressure of 1 kbar accelerates the low-temperature tunnelling process by a factor of 2. [Mn1−xFex(bpy)3](PF6)2 and [Zn1−xFex(bpy)3](PF6)2show a phase transition at ≈1.1 kbar, which increases the tunnelling rate by a factor of about 6. | ||||||||
|
||||||||
The thermal spin transition in the diluted mixed crystal [Zn1−xFex(6-mepy)3tren](PF6)2 (x = 0.00025, (6-mepy)3tren = tris{4-[(6-methyl)-2-pyridyl]-3-aza-3-butenyl}amine) is studied at 1 bar and 1 kbar by temperature-dependent absorption spectroscopy. From thermodynamic analysis of the high-spin (HS) fractions, values for ΔHHL0 and ΔSHL0 of 1551(50) cm−1 and 7.5(5) cm−1/K and a molecular volume of reaction, ΔVHL0, of 22(2) Å3result. Reconsideration of the cooperative effects in the neat [Fe(6-mepy)3tren](PF6)2from Adler et al. [Hyperfine Interact. 47, 343 (1989)] result in a lattice shift, Δ, of 208(15) cm−1 and an interaction constant, Γ, of 109(15) cm−1. Temperature-dependent laser flash photolysis experiments in the spin-crossover system [Zn1−xFex(6-mepy)3tren](PF6)2 and the LS system [Zn1−xFex(py)3tren](PF6)2 in the pressure range between 1 bar and 1 kbar are presented. Above ≈100 K the HS→LS (low-spin) relaxations behave classically, whereas they become almost temperature independent below 50 K. At ambient pressure, the low-temperature tunneling rate constant in[Zn1−xFex(py)3tren](PF6)2 is more than three orders of magnitude larger than the one in[Zn1−xFex(6-mepy)3tren](PF6)2. External pressure of 27 kbar accelerates the low-temperature tunneling process by almost nine orders of magnitude. The kinetic results are discussed within the theory of nonadiabatic multiphonon relaxation. | ||||||||
|
||||||||
The spin-crossover compound [Fe(pic)3]Cl2EtOH (pic = 2-picolylamine) shows an unusual two-step spin transition. This is thought to be caused by specific nearest-neighbour interactions and short-range correlations and requires a theoretical treatment of the elastic interactions between the spin-changing molecules beyond the mean-field approximation. Such short-range correlations also influence the high-spin → low-spin relaxation following the light-induced population of the high-spin state at cryogenic temperatures, leading to characteristic deviations from the predictions of a mean-field treatment. These deviations are directly observable by comparison of the full and unperturbed relaxation curves with curves for which the short-range correlations were destroyed using an appropriate irradiation technique. Monte Carlo simulations including both nearest-neighbour and long-range interactions give a description of the observed relaxation curves which is consistent with the thermal spin equilibrium. | ||||||||
|
||||||||
Polymeric two- and three-dimensional, homo- and heterometallic oxalatebridged coordination compounds offer exciting opportunities, mainly in the fields of molecular magnetism and photophysics. Given that a large variety of magnetic phenomena have been reported so far from these molecular magnets, very limited experience is gained from elastic neutron scattering experiments. Therefore, with two examples, we will address the topic of the elucidation of magnetic structures by means of the neutron scattering technique. In addition, due to the possibility of the variation of different metal ions in varying oxidation states, interesting photophysical processes can be observed within the extended three-dimensional host/guest systems. | ||||||||
|
||||||||
A helium gas pressure cell for pressures up to 1 kbar (0.1 GPa) has been developed in conjunction with a closed-cycle He refrigerator allowing variable temperatures between 15 and 300 K. Both cell and refrigerator are equipped with optical windows suitable for photophysical measurements, such as temperature- and pressure-dependent absorption spectroscopy or laser flash photolysis. Examples of measurements on iron(II) spin-crossover systems are given. In these compounds, comparatively small external pressures induce significant changes in the thermodynamic equilibrium as well as in the relaxation dynamics. | ||||||||
|
 |
|||||||
Based on a synthetic strategy, extended anionic, homo and bimetallic oxalato-bridged transition-metal compounds with two (2D) and three-dimensional (3D) connectivities can be synthesized and crystallized. Thereby, the choice of the templating counterions will determine the crystal chemistry. Since the oxalato bridge is a mediator for both antiferro and ferromagnetic interactions between similar and dissimilar metal ions, long-range magnetic ordering will occur. Examples of the determination of magnetic structures in 2D and 3D compounds by means of elastic neutron scattering methods will be discussed. In addition, due to the possibility of the variation of different metal ions in varying oxidation states, interesting photophysical processes can be observed within the extended three-dimensional host/guest systems. | ||||||||
|
||||||||
Laser flash photolysis experiments were performed on the mixed crystal [Zn1−xFex(6-mepy)3tren](PF6)2 (x=0.00025) at 10 K in the pressure range between 1 bar and 20 kbar. An external pressure of 20 kbar accelerates the low-temperature tunneling process by almost eight orders of magnitude. | ||||||||
|
||||||||
The iron(II) spin-crossover compound [Fe(ptz)6](PF6)2 (ptz = 1-propyltetrazole) crystallizes in the triclinic space group P†, with a = 10.6439(4) Å, b = 10.8685(4) Å, c = 11.7014(4) Å, α = 75.644(1)°, β = 71.671(1)°, γ = 60.815(1)°, and Z = 1. In [Fe(ptz)6](PF6)2, the thermal spin transition is extremely steep because of cooperative effects of elastic origin. The transition temperature at ambient pressure is 74(1) K. An external pressure of 1 kbar shifts the transition temperature to 102(1) K, corresponding to a stabilization of the low-spin state, which is smaller in volume. The volume difference between the high-spin and the low-spin state, ΔV°HL, is 24(2) Å3/molecule. The interaction constant Γ, as a measure of cooperativity, is within experimental error independent of external pressure and has a value of 101(5) cm-1. In contrast to the case of the related compound [Fe(ptz)6](BF4)2 (Decurtins et al. Inorg. Chem. 1985, 24, 2174), there is no hysteresis due to a first-order crystallographic phase transition, nor is there a hysteresis induced by external pressure as in the mixed crystal [Zn1-xFex(ptz)6](BF4)2, x = 0.1 (Jeftić et al. J. Phys. Chem. Solids 1996, 57, 1743). However, in [Fe(ptz)6](PF6)2, the interaction constant Γ is found to be very close to the critical value above which a hysteresis solely due to the cooperative effects is expected. In addition, high-spin → low-spin relaxation measurements were performed under external pressures of up to 1 kbar in the temperature interval between 50 and 60 K. An external pressure of 1 kbar accelerates the high-spin → low-spin relaxation by 1 order of magnitude. | ||||||||
|
||||||||
In the iron(II) spin-crossover compound [Fe(ptz)6](BF4)2, the thermal spin transition is accompanied by a crystallographic phase transition showing a hysteresis with Tc↓ = 128 K and Tc↑ = 135 K at ambient pressure [Franke, P. L.; Haasnot, J. G.; Zuur, A. P. Inorg. Chim. Acta 1982, 59, 5]. The hysteresis is due to an interplay between the spin-transition and the R3 → P† crystallographic phase transition with a large low-spin fraction stabilizing the P† phase at low temperatures. In the mixed crystal [Zn1-xFex(ptz)6](BF4)2, x = 0.1, with the iron complexes imbedded into the isomorphous zinc lattice, the crystallographic phase transition can be induced by an external pressure [Jeftić, J.; Romstedt, H.; Hauser, A. J. Phys. Chem. Solids 1996, 57, 1743]. Thus the P† phase is additionally stabilized by external pressure. The interaction constant Γ, which describes cooperative effects between the spin-changing complexes, differs for the two crystallographic phases. Values for Γ(P†) of 144(8) cm-1 and the volume difference ΔV0HL of 29(4) Å3 are determined from a simultaneous fit to a series of transition curves for different pressures and iron content x in the P† phase. These values are compared to the corresponding values for the R3 phase, viz. Γ(R3) of 170(9) cm-1 and ΔV0HL(R3) of 26(3) Å3. Surprisingly Γ(R3) is larger than Γ(P†) despite the fact that ΔV0HL(R3) is smaller than ΔV0HL(P1). The high-spin → low-spin relaxation at temperatures above ~80 K is thermally activated, while below ~40 K temperature independent tunnelling takes place. An external pressure of 1 kbar accelerates the high-spin → low-spin relaxation exponentially by 1 order of magnitude in the tunnelling region in both crystallographic phases and regardless of x. In the concentrated material the high-spin → low-spin relaxation is self-accelerating due a buildup of an internal pressure [Hauser, A. Chem. Phys. Lett. 1992, 192, 65]. Both cooperative effects and external pressure result in a shift of the maximum of the 1A1 → 1T1 absorption band. | ||||||||
|
||||||||
A detailed analysis of Zeeman splittings of highly resolved spin-forbidden transitions in [Cr(bpy)3](PF6)3 is presented. Assignments of vibronic bands are made based on low-temperature absorption, emission, and infrared spectra. The pattern of doublet states, obtained for H = 0 and H = 5 T, is consistent with angular overlap model (AOM) calculations, which allow one to consider σ- and π-interactions between the metal-d and relevant ligand orbitals and the particular angular geometry of the chromophore simultaneously. The observed level splittings are found to result from the combined effect of trigonal distortion and contributions of the symmetry adapted dπ-orbitals involved due to coupling with corresponding counterparts from the bidentate ligand (phase coupling). The larger splitting of the lowest excited state 2Eg(Oh) in the analogous ClO4- salt is due to the more distorted geometry of the [CrN6] moiety. Related properties of the bipyridine ligand, which turn out to show donor behavior in the present compounds, and the acetylacetonate ligand are discussed, and AOM parameters for the metal−ligand π-interaction are correlated with results of MO calculations. | ||||||||
|
||||||||
Resonant fluorescence line narrowing of the R1 line of the [Cr(ox)3]3− chromophore in [Rh(bpy)3][NaCr(ox)3]ClO4 at 1.6 K neither gives rise to the usual three-line pattern nor to spectral diffusion. Instead multi-line spectra with spacings equal to the zero-field splitting of the ground state are observed. This phenomenon is attributed to efficient non-radiative resonant energy transfer within the R1 line. | ||||||||
|
||||||||
In resonant fluorescence line narrowing (FLN) experiments in the R1 transition of the [Cr(ox)3]3- chromophore in [Ru(bpy)3][NaAl:Cr(1%)(ox)3] and [Rh(bpy)3][NaCr(ox)3]ClO4 multiline spectra are observed at 1.8 K, (ox=oxalate, bpy=2,2’-bipyridine). For [Rh(bpy)3][NaCr(ox)3]ClO4 the number of lines and their relative intensities depend critically upon the excitation wavelength within the inhomogeneous distribution, and in time-resolved FLN experiments additionally upon the delay. This behavior is clear evidence for a resonant energy-transfer process. At 4.2 K the more common phonon-assisted process becomes dominant, manifesting itself as spectral diffusion. | ||||||||
|
||||||||
Chemical variation and combination of metal ions of different valencies in the oxalate backbone as well as in the tris-bpy cation of the three-dimensional network structures of the type [MII2(ox)3][MII(bpy)3] (bpy = 2,2'-bipyridine, ox = C2O42-), [MIMIII(ox)3][MII(bpy)3] and [MIMIII(ox)3][MIII(bpy)3]ClO4 offer unique opportunities for studying a large variety of photophysical processes. Depending upon the relative energies of the excited states of the chromophores, excitation energy transfer either from the tris-bipyridine cation to the oxalate backbone or vice versa is observed, as for instance from [Ru(bpy)3]2+ as photo-sensitiser to [Cr(ox)3]3- as energy acceptor in the combination [NaCr(ox)3][Ru(bpy)3], or from [Cr(ox)3]3- to [Cr(bpy)3]3+ in [NaCr(ox)3][Cr(bpy)3]ClO4. In addition efficient energy migration within the oxalate backbone is observed. Furthermore, depending upon the excited state redox potentials, light-induced electron transfer processes may be envisaged. | ||||||||
|
||||||||
In analogy to the [MII(bpy)3]2+ cations, where MII is a divalent transition-metal and bpy is 2,2‘-bipyridine, the tris-chelated [MIII(bpy)3]3+ cations, where MIII is CrIII or CoIII, induce the crystallization of chiral, anionic three-dimensional (3D) coordination polymers of oxalate-bridged (μ-ox) metal complexes with stoichiometries [MII2(ox)3]n2n- or [MIMIII(ox)3]n2n-. The tripositive charge is partially compensated by inclusion of additional complex anions like ClO4-, BF4-, or PF6- which are encapsulated in cubic shaped cavities formed by the bipyridine ligands of the cations. Thus, an elaborate structure of cationic and anionic species within a polymeric anionic network is realized. The compounds isolated and structurally characterized include [CrIII(bpy)3][ClO4] [NaCrIII(ox)3] (1), [CrIII(bpy)3][ClO4][MnII2(ox)3] (2), [CrIII(bpy)3][BF4] [MnII2(ox)3] (3), [CoIII(bpy)3][PF6][NaCrIII(ox)3] (4). Crystal data: 1, cubic, P213, a = 15.523(4) Å, Z = 4; 2, cubic, P4132, a = 15.564(3) Å, Z = 4; 3, cubic, P4132, a = 15.553(3) Å, Z = 4; 4, cubic, P213, a= 15.515(3) Å, Z = 4. Furthermore, it seemed likely that 1,2-dithiooxalate (dto) could act as an alternative to the oxalate bridging ligand, and as a result the compound [NiII(phen)3][NaCoIII(dto)3]·C3H6O (5) has successfully been isolated and structurally characterized. Crystal data: 5, orthorhombic, P212121, a = 16.238(4) Å, b = 16.225(4) Å, c = 18.371(5) Å, Z = 4. In addition, the photophysical properties of compound 1 have been investigated in detail. In single crystal absorption spectra of [CrIII(bpy)3][ClO4][NaCrIII(ox)3] (1), the spin−flip transitions of both the [Cr(bpy)3]3+ and the [Cr(ox)3]3- chromophores are observed and can be clearly distinguished. Irradiating into the spin-allowed 4A2 → 4T2absorption band of [Cr(ox)3]3- results in intense luminescence from the 2E state of [Cr(bpy)3]3+as a result of rapid energy transfer processes. | ||||||||
|
||||||||
The high-spin → low-spin relaxation dynamics of the Fe(III) spin-crossover complexes [Fe(Sal2tr)]PF6 (H2Sal2tr = Bis(salicylaldimino)triethylenetetramine) and [Fe(acpa)2]PF6(Hacpa = N-(1-acetyl-2-propylidene)-2-pyridylmethylamine) are discussed within the theory of nonadiabatic multiphonon relaxation. A Huang−Rhys factor S of ≈25, estimated on the basis of average metal−ligand bond length differences ΔrHL of ≈ 0.12 Å, explains the observed low-temperature tunneling rate constants kHL(T→0) of ≈ 102 s-1 as well as the thermally activated process at T > ≈100 K semiquantitatively. The results obtained for the Fe(III) compounds are compared to those for Fe(II) spin-crossover compounds. | ||||||||
|
||||||||
At low temperatures an external pressure of 1 kbar accelerates the high-spin → low-spin relaxation in the [Zn1−xFex(ptz)6](BF4)2, X = 0.1, spin-crossover system by one order of magnitude. This is due to the large difference in volume between the high-spin and low-spin states of 26 Å3/molecule. The relative vertical and horizontal shifts of the potential wells of the two states as a function of pressure are estimated to be 130 cm−1/kbar and 10−3 Å/kbar, respectively. | ||||||||
|
||||||||
In the [Fe(ptz)6](BF4)2 (ptz = 1-propyltetrazole) spin-crossover system, the thermal spin transition is accompanied by a first order crystallographic phase transition (Tc↓ = 128 K and Tc↑ = 135 K) from R3 above Tc↓ to P1 at low temperatures (Wiehl L., Acta Cryst. B49, 289 (1993)). The high-symmetry phase can be super-cooled, in which case the spin transition is still complete and quite steep (T1/2 = 125 ± 2 K) but now without a hysteresis. The corresponding interaction constant Γ is 170 cm−1. In the diluted system [Zn1−xFex(ptz)6](BF4)2, X = 0.1, the spin transition is gradual with T1/2 = 95 ± 2 K. From the shift of T1/2 towards high temperatures with external pressure a value for ΔVHL0 of 26 Å3 molecule−1 is obtained. Pressures above 250 bar induce a crystallographic phase transition even in the diluted system, as a result of which the spin transition is discontinuous. The interplay between the thermal spin transition and the crystallographic phase transition in the neat and the diluted system is discussed consistently. | ||||||||
|
||||||||
In the [Fe(etz)6](BF4)2 spincrossover system the iron(II) complexes occupy two nonequivalent lattice sites, sites A and B. Complexes on site A show a thermal high-spin (HS) → low-spin (LS) transition at 105 K, whereas complexes on site B remain in the HS state down to 10 K. Complexes on both sites exhibit light-induced spin state conversions (LIESST) at 20 K: LS → HS on site A with λ = 514.5 nm, and HS → LS on site B with λ = 820 nm. The relaxation processes subsequent to the HS → LS conversion on site B reveal a light-induced HS→LS bistability for the complexes on site B at 70 K. The bistability as well as the absence of a thermal spin transition on site B are attributed to a thermal hysteresis for the B-site complexes with a critical temperature T↑c K on heating. This hysteresis can be interpreted in terms of strong cooperative effects of elastic origin, which, in addition, cause characteristic deviations of the relaxation on site B from first-order kinetics (self-acceleration). In contrast, the HS → LS relaxation at 60 K on site A after irradiation with λ = 514.5 nm shows an unusual self-retardation. | ||||||||
|
||||||||
The [Fe(etz),](BF,), spin-cross-over system (etz = 1-ethyl-1 H-tetrazole) crystallizes in space group P1, with the following lattice constants at 298 K: a10.419(3), b=15.709(1), c = 18.890(2) Å, α = 71.223(9), β =77.986(10), and γ = 84.62(1)° V = 2862.0(9) Å3 and Z = 3. Two nonequivalent lattice sites, one without (site A) and one with (site B) inversion symmetry, are observed. The population of the two sites nA:nB is 2:l. Iron(II) on site A undergoes a thermal low-spin (LS) → high-spin (HS) transition with T1/2I, = 105 K. whereas that on site B remains in the high-spin state down to cryogenic temperatures. Application of external pressure of up to 1200 bar between 200 and 60 K does not cause formation of the low-spin state on site B. On site A the high-spin state can be populated as a metastable state at 20 K by irradiating the sample with λ = 514.5 nm; on site B a light-induced population of the low-spin state can be achieved with λ = 820 nm. | ||||||||
|
||||||||
Intersystem crossing is the crucial first step determining the quantum efficiency of very many photochemical and photophysical processes. Spin-crossover compounds of first-row transition metal ions, in particular of Fe(II), provide model systems for studying it in detail. Because in these compounds there are no competing relaxation processes, intersystem crossing rate constants can be determined over a large temperature interval. The characteristic features are tunnelling at temperatures below ∼80 K and a thermally activated process above ∼ 100 K. This, as well as the twelve order of magnitude increase of the low-temperature tunnelling rate constant on going from a spin-crossover compound with a small zero-point energy difference to a low-spin compound with a substantially larger one, can be understood on the basis of a nonadiabatic multiphonon process in the strong vibronic coupling limit. | ||||||||
|
||||||||
The absorption spectra of the ferrate (VI) ion (FeO2-4) in K2MO4 (M = S, Se, Cr, Mo) host lattices consist of a series of relatively weak bands at low energy, which can be assigned to transitions within the partially filled 3d shell and some intense bands at higher energy, which are assigned to ligand-to-metal charge transfer transitions (LMCT). In the near-infrared (NIR) region sharp lines are observed belonging to the spin-forbidden spin-flip transitions 3A2→ 1E and 3A2 → 1A1. The lowest excited state is the 1E state, serving as initial state for 1E → 3A2 sharp-line luminescence at around 6200 cm-1. Another luminescence is observed centered at 9000 cm-1, which is assigned to the 3T2 → 3A2 transition. It is rather broad and three orders of magnitude weaker than the 1E luminescence at 30K as a result of efficient non-radiative relaxation processes to the 1E state. The temperature dependence of the total intensity and the lifetime of the 1E → 3A2 luminescence is understood within a complex scheme of radiative and non-radiative processes. | ||||||||
|
|
|
||||||||
The mixed-metal ferromagnet {[P(Ph)4][MnCr(ox)3]}n, where Ph is phenyl and ox is oxalate, has been prepared and a two-dimensional network structure, extended by Mn(II)-ox-Cr(III) bridges, has been determined from single crystal X-ray data. Crystal data: space group R3c, a=b=18.783(3), c=57.283(24) Å, α=β=90, γ=120°, Z=24 (C30H20O12PCrMn). The magnetic susceptibility data obey the Curie-Weiss law in the temperature range 260–20 K with a positive Weiss constant of 10.5 K. The temperature dependence of the molar magnetization exhibits a magnetic phase transition at Tc=5.9 K. The structure is discussed in relation to the strategy for preparing molecular based ferromagnets and, in addition, it is a solution to the question of the dimensionality of the [MM'(ox)3]n network, which in principle can extend two- or three-dimensionally to the crystal lattice. The optical absorption spectra of the single crystals are assigned to the ‘CrO6' chromophores. Their polarization patterns reflect the electric dipole selection rules for D3 symmetry. A strong site selective luminescence from the chromium(III) 2E states is observed at low temperature and the system may be suitable for studying energy transfer mechanisms. | ||||||||
|
|
||||||||
Transition metal chemistry contains a class of complex compounds for which the spin state of the central atom changes from high spin to low spin when the temperature is lowered. This is accompanied by changes of the magnetic and optical properties that make the thermally induced spin transition (also called spin crossover) easy to follow. The phenomenon is found in the solid state as well as in solution. Amongst this class, iron(II) spin crossover compounds are distinguished for their great variety of spin transition behavior; it can be anything from gradual to abrupt, stepwise, or with hysteresis effects. Many examples have been thoroughly studied by Mössbauer and optical spectroscopy, measurements of the magnetic susceptibilities and the heat capacities, as well as crystal structure analysis. Cooperative interactions between the complex molecules can be satisfactorily explained from changes in the elastic properties during the spin transition, that is, from changes in molecular structure and volume. Our investigations of iron(II) spin crossover compounds have shown that green light will switch the low spin state to the high spin state, which then can have a virtually unlimited lifetime at low temperatures (this phenomenom is termed light-induced excited spin state trapping - acronym: LIESST). Red light will switch the metastable high spin state back to the low spin state. We have elucidated the mechanism of the LIESST effect and studied the deactivation kinetics in detail. It is now well understood within the theoretical context of radiationless transitions. Applications of the LIESST effect in optical information technology can be envisaged. | ||||||||
|
||||||||
In der Übergangsmetallchemie gibt es eine Klasse von Komplexverbindungen, bei denen eine Temperaturerniedrigung einen Wechsel im Spinzustand des Zentralatoms vom High-Spin- in den Low-Spin-Zustand bewirkt. Dabei ändern sich die magnetischen und optischen Eigenschaften, über die der thermische Spinübergang (auch Spincrossover genannt) sehr gut verfolgt werden kann. Dieses Phänomen tritt sowohl in flüssiger Phase als auch im Festkörper auf. Eine herausragende Stellung nehmen Eisen(II) - Spincrossover - Verbindungen ein, in denen der Spinübergang im Festkörper auf sehr unterschiedliche Weise - graduell, abrupt, mit Hysterese oder stufenweise - verlaufen kann und mit Mößbauer- und optischer Spektroskopie, mit magnetischen Suszeptibilitäts- und Wärmekapazitätsmessungen sowie durch Kristallstrukturanalyse intensiv untersucht worden ist. Die kooperative Wechselwirkung zwischen den einzelnen Komplexmolekülen kann befriedigend durch elastische Eigenschaften und durch die Änderung von Gestalt und Volumen der Komplexmoleküle beim Spinübergang erklärt werden. Bei Untersuchungen an Eisen(II)-Spincrossover-Verbindungen konnte man beobachten, daß sich der Low-Spin-Zustand mit grünem Licht in den High-Spin-Zustand umschalten läßt, der bei tiefen Temperaturen eine nahezu unendlich lange Lebensdauer haben kann (LIESST = Light-Induced Excited Spin State Trapping). Mit rotem Licht läßt sich der metastabile High-Spin- wieder in den Low-Spin-Zustand zurückschalten. Der Mechanismus des LIESST-Effekts ist aufgeklärt, die Zerfallskinetik im Detail untersucht und im Rahmen der Theorie strahlungsloser Übergänge verstanden. Anwendungen des LIESST-Effekts in der optischen Informationstechnik sind denkbar. | ||||||||
|
||||||||
The well-resolved absorption, excitation, and luminescence spectra of [Ir(ppy)2bpy]+ (ppyH = 2-phenylpyridine, bpy = 2,2'-bipyridine) in different media at cryogenic temperatures are presented. In solutions and glasses at ambient temperature the lowest energy excited state corresponds to an Ir - bpy charge-transfer excitation whereas in the crystalline host lattice [Rh(ppy)2bpy]PF6 the lowest excited state at 21 450 cm-1 is assigned to a 37r-r* excitation localized on the cyclometalating ppy- ligands. The next higher excited Ir - bpy charge-transfer state has shifted to 21 820 cm-', only 300 cm-I above the 3LC excited state. The close proximity of the 3LC and 3MLCT excited states and the large spin-orbit coupling constant of Ir3+ induce a strong mixing of charge-transfer character into the 3LC lowest excited states, resulting in increased oscillator strengths, reduced lifetimes, short axis polarized transitions, and a large zero-field splitting of 10-15 cm-1. | ||||||||
|
||||||||
Making use of the phenomenon of light-induced spin-crossover in the [Zn1−xFex(ptz)6](BF4)2 spin-crossover system, very high diffraction efficiencies η can be achieved in non-degenerate four-wave-mixing. In the mixed crystal with x=0.1 and at 76 K, i.e. at a temperature where the system is predominantly in the low-spin state, a value for η of 43% was obtained. This is attributed to a phase grating due to the large difference in metal---ligand bondlength between the low-spin ground state and the light-induced high-spin state. The rate constant for the decay of the laser-induced grating as a function of temperature is found to be exactly twice the one of the high-spin→low-spin relaxation, as expected for a dilute system in the absence of cooperative effects. | ||||||||
|
||||||||
The two title compounds were synthesized and investigated with the inelastic-neutron-scattering (INS) technique. They contain mixed YbMBr93- (M=Cr3+, Ho3+) dimers as discrete units, and the magnetic excitations of mixed Yb3+-Cr3+ and Yb3+-Ho3+ dimers could thus be observed. The Yb3+-Cr3+ dimer has three INS transitions, for which anisotropic exchange, as well as zero-field splitting of Cr3+, has to be included in the exchange Hamiltonian. For the Yb3+-Ho3+ dimer the effect of the exchange interaction manifests itself as a broadening and a splitting of the crystal-electric-field levels of the isolated Ho3+ ion. Taking into account the full (2J + 1) ground-state multiplet of Ho3+, as well as anisotropic exchange, gives a satisfactory description of this dimer. | ||||||||
|
||||||||
The high-spin to low-spin (HS→LS) relaxation in the [Fe(ptz)6](BF4)2 spin-crossover system deviates strongly from first-order kinetics because of cooperative effects of elastic origin. The shift in horizontal and vertical displacement of the potential wells of the initial and final state relative to each other due to the build-up of an "internal" pressure is estimated from spectroscopic measurements. The HS→LS relaxation as such is described by the theory of nonadiabatic multiphonon relaxation in the strong-coupling limit, with a Huang—Rhys factor S ≈ 45 which is much larger than the reduced energy gap p. The sigmoidal relaxation curves in [Fe(ptz)6](BF4)2 result when a change in S of ≈ −1 and in p of 1 during the relaxation is taken into account. | ||||||||
|
||||||||
The 5T2(HS)1A1(LS) intersystem crossing rates have been determined for a number of Fe(II) coordination compounds between 10 and 270 K using time-dependent optical spectroscopy. Strong deviations from Arrhenius kinetics with nearly temperature independent tunneling at low temperatures and a thermally activated behavior at elevated temperatures with apparent activation energies smaller than the classical energy barrier were found. The tunneling rates range from ~10−6 s−1 for the doped spin crossover system [Zn1−xFex(ptz)6](BF4)2 to ~106 s−1 for the doped low-spin (LS) system [Zn1−xFex(bipy)3](PF6)2. The large range of 12 orders of magnitude in the low temperature tunneling rates as well as the activated region can be understood in terms of nonadiabatic multiphonon relaxation. Values for the Huang–Rhys parameter S of 40–50 and for the reduced energy gap p of 1–12 are estimated for the present series of compounds. The validity of an inverse energy gap law in the strong vibronic coupling limit with Sp is borne out by experiment. | ||||||||
|
||||||||
Due to the fact that for d6systems there are a number of low-lying ligand field (LF) states the relaxation from excited states of Fe(II) coordination compounds is, in general, a very fast and radiationless process. In Fe(II) spin-crossover systems, however, the zero point energy difference between the two lowest states, namely the low-spin (LS) 1A1 and the high-spin (HS) 5T2 state, is of the order of kBT, and some systems can be converted quantitatively to the HS state well below the thermal transition temperature by irradiating either into MLCT or LF absorption bands of the LS species, with HS→LS relaxation rates as small as 10−6 s−1 at XXX10 K. It is also possible to achieve a light-induced transient population of a HS state in Fe(II) LS compounds, but in this case the HS→LS relaxation rates can be larger than 106 s−1 even at low temperatures. The HS→LS relaxation rates show strong deviations from Arrhenius kinetics with nearly temperature independent tunnelling below ˜70 K and a thermally activated behaviour above ˜100 K. The range of 12 orders of magnitude in the low temperature tunnelling rate can be understood in terms of nonadiabatic multiphonon relaxation, where in the strong coupling limit, with the Huang-Rhys parameter S much larger than the reduced energy gap p, an inverse energy gap law holds. | ||||||||
|
||||||||
[Fe(ptz)6](BF4)2 (ptz=1-propyltetrazole) is an Fe(II) spin crossover system, which shows a light-induced low-spin (1A1)-->high-spin (5T2) conversion below ~50 K by irradiating into the spin allowed 1A1-->1T1 d–d absorption band. This phenomenon, known as light-induced excited spin state trapping (LIESST), is reversible, and a subsequent irradiation into the 5T2-->5E band results in a light-induced 5T2-->1A1 conversion (reverse LIESST). Single crystal absorption spectra of the title compound in the region of d–d transitions are reported. In addition to the well-established spin allowed 1A1-->1T1 and 1A1-->1T2 transitions of the low-spin species and the 5T2-->5E transition of the high-spin species two weak bands in the NIR are assigned to the spin forbidden 1A1-->3T1 and 1A1-->3T2 transitions. Direct irradiation into the 1A1-->3T1 absorption band at 20 K results in a quantitative 1A1-->5T2 conversion, proving that this low lying triplet state plays an important role in the mechanism of LIESST. A full kinetic scheme for LIESST and reverse-LIESST with the 3T1 state as intermediate state is developed, and the quantum efficiencies for the various intersystem crossing steps involved are given: they are of the order of unity for the first step from the initially excited 1T1 and 5E states to the intermediate 3T1 state, respectively. The branching ratio from the 3T1 state to the 1A1 and the 5T2 states is 1:4. | ||||||||
|
||||||||
Recently, we have discovered a fascinating photophysical effect in spin crossover complexes of iron(II) : Light-Induced Excited Spin State Trapping (LIESST). At sufficiently low temperatures, the low spin state (1A1) can be converted quantitatively to the high spin state (5T2) by irradiating the sample into the 1A1 → 1T1 d-d absorption band (—540 nm). The resulting metastable HS state has a very long lifetime at low temperatures, in some cases it does not decay noticeably over a period of several days at 10 K. Only at temperature above some critical temperature does thermal relaxation back to the LS state set in. The sample can also be reconverted to the LS state by irradiating into the 5T2 → 5E absorption band (∞50 nm). The system thus behaves like an optical switch. The relative positioning - horizontally and vertically - of the potential wells of the two spin states is crucial for the lifetime of the metastable HS state. | ||||||||
|
||||||||
It is known that [Fe (2-mephen)3] (ClO4)2 in the solid state is an iron(II) spin-crossover system which shows light-induced excited spin state trapping (LIESST). The thermal spin-crossover behaviour of the complex [Fe(2-mephen)3]2+ embedded in various polymer matrices is similar to the solid-state behaviour, and a light-induced long-lived excited state is observed at temperatures below 50 K. Relaxation curves show that polymer matrices are not very homogeneous media. | ||||||||
|
||||||||
Fe(ptz)6(BF4)2 (ptz = 1-propyltetrazole) is an iron(II) spin-crossover system which shows light-induced excited spin state trapping. In this paper we show that (a) the same phenomenon can also be observed in Zn1−xFex(ptz)6(BF4)2 (x ≈ 0.1) and is therefore basically a single-ion property, and (b) that the phenomenon is reversible. The efficiency of the light-induced spin crossover is of the order of 0.5% in the forward direction and 0.1% in the reverse direction. | ||||||||
|
||||||||
[Fe(ptz)6l(BF4)2 (ptz= 1-propyltetrazole) and the mixed crystals [Znl,Fe,(ptz)6] (BF4)2 are Fe(I1) spin-crossover compoundsthat exhibit light-induced excited-spin-state trapping. It is shown that (a) for x I 0.1 a single-ion treatment of both the spinequilibrium ( M H L = 510 (12) cm-', ASHL = 5.1 (2) cm-'/K at T = 100 K) and the relaxation from the excited high-spin state (.Eao = 810 (30) cm-I, A - 105/s) is appropriate and (b) for 0.2 I x I 1 cooperative effects observed in the relaxation from the high-spin state are of long-range nature and therefore of elastic rather than of electronic origin. | ||||||||
|
||||||||
Low-temperature luminescence and absorption spectra were recorded of VClz doped into MgCl, and CdCl, as well as of the pure compound. There is evidence for excitation energy transfer in Velz down to 5 K. In the diluted materials the luminescence remains unquenched up to 200 K (Cd,,V,Cl2) and 250 K (MgI-,V,CI2). The broad-band 4T2, - 4Azs luminescence transition is highly structured in the diluted samples. There is multiple evidence for a Jahn-Teller effect in the 4Tz, state with an estimated Jahn-Teller energy of the order of 200 cm-I. Polarized absorption and Zeeman meausurements were used to assign the 4Az, -,E,, *TI, transitions. | ||||||||
2017
2016
2015
2014
2013
2012
2011
2010
2009
2008
2007
2006
2005
2004
2003
2002
2001
2000
1999
1998
1997
1996
1995
1994
1993
1992
1991
1990
1988
1986
1985
Download this list in format RIS
 EndNote
EndNote  BibTex
BibTex  PDF XML
PDF XML Last update Friday December 08 2017